

Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics
- PMID: 33805567
- PMCID: PMC8037620
- DOI: 10.3390/ijms22073553
Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics
Abstract
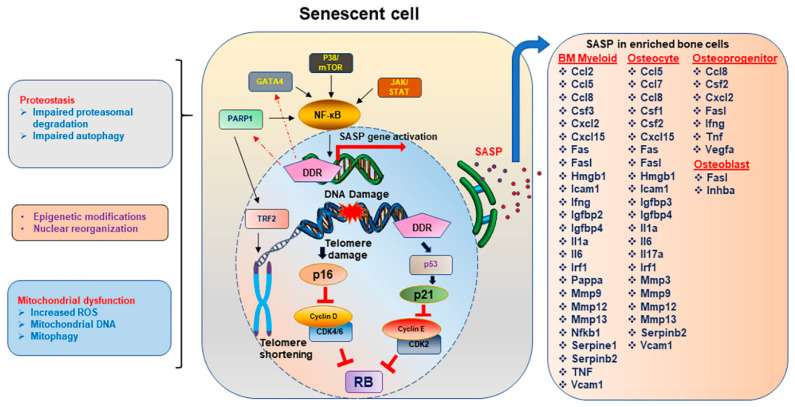
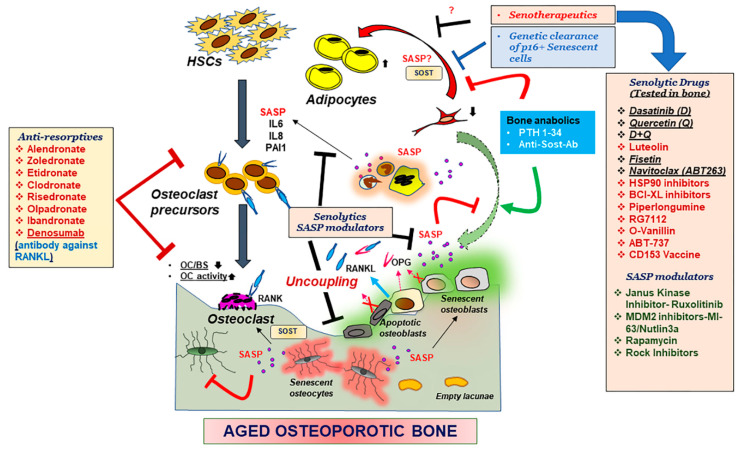
Bone is a dynamic organ maintained by tightly regulated mechanisms. With old age, bone homeostasis, which is maintained by an intricate balance between bone formation and bone resorption, undergoes deregulation. Oxidative stress-induced DNA damage, cellular apoptosis, and cellular senescence are all responsible for this tissue dysfunction and the imbalance in the bone homeostasis. These cellular mechanisms have become a target for therapeutics to treat age-related osteoporosis. Genetic mouse models have shown the importance of senescent cell clearance in alleviating age-related osteoporosis. Furthermore, we and others have shown that targeting cellular senescence pharmacologically was an effective tool to alleviate age- and radiation-induced osteoporosis. Senescent cells also have an altered secretome known as the senescence associated secretory phenotype (SASP), which may have autocrine, paracrine, or endocrine function. The current review discusses the current and potential pathways which lead to a senescence profile in an aged skeleton and how bone homeostasis is affected during age-related osteoporosis. The review has also discussed existing therapeutics for the treatment of osteoporosis and rationalizes for novel therapeutic options based on cellular senescence and the SASP as an underlying pathogenesis of an aging bone.
Keywords: SASP; aging; osteoporosis; radiation; senescence; senotherapeutic.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Riggs B.L., Khosla S., Melton L.J., 3rd A unitary model for involutional osteoporosis: Estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J. Bone Miner. Res. 1998;13:763–773. doi: 10.1359/jbmr.1998.13.5.763. - DOI - PubMed
-
- Farr J.N., Rowsey J.L., Eckhardt B.A., Thicke B.S., Fraser D.G., Tchkonia T., Kirkland J.L., Monroe D.G., Khosla S. Independent Roles of Estrogen Deficiency and Cellular Senescence in the Pathogenesis of Osteoporosis: Evidence in Young Adult Mice and Older Humans. J. Bone Miner. Res. 2019;34:1407–1418. doi: 10.1002/jbmr.3729. - DOI - PMC - PubMed
-
- Khosla S., Pacifici R. Chapter 46–Estrogen Deficiency, Postmenopausal Osteoporosis, and Age-Related Bone Loss. In: Marcus R., Feldman D., Dempster D.W., Luckey M., Cauley J.A., editors. Osteoporosis. 4st ed. Academic Press; San Diego, CA, USA: 2013. pp. 1113–1136.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
