

A Comprehensive Review of Genetically Engineered Mouse Models for Prader-Willi Syndrome Research
- PMID: 33807162
- PMCID: PMC8037846
- DOI: 10.3390/ijms22073613
A Comprehensive Review of Genetically Engineered Mouse Models for Prader-Willi Syndrome Research
Abstract
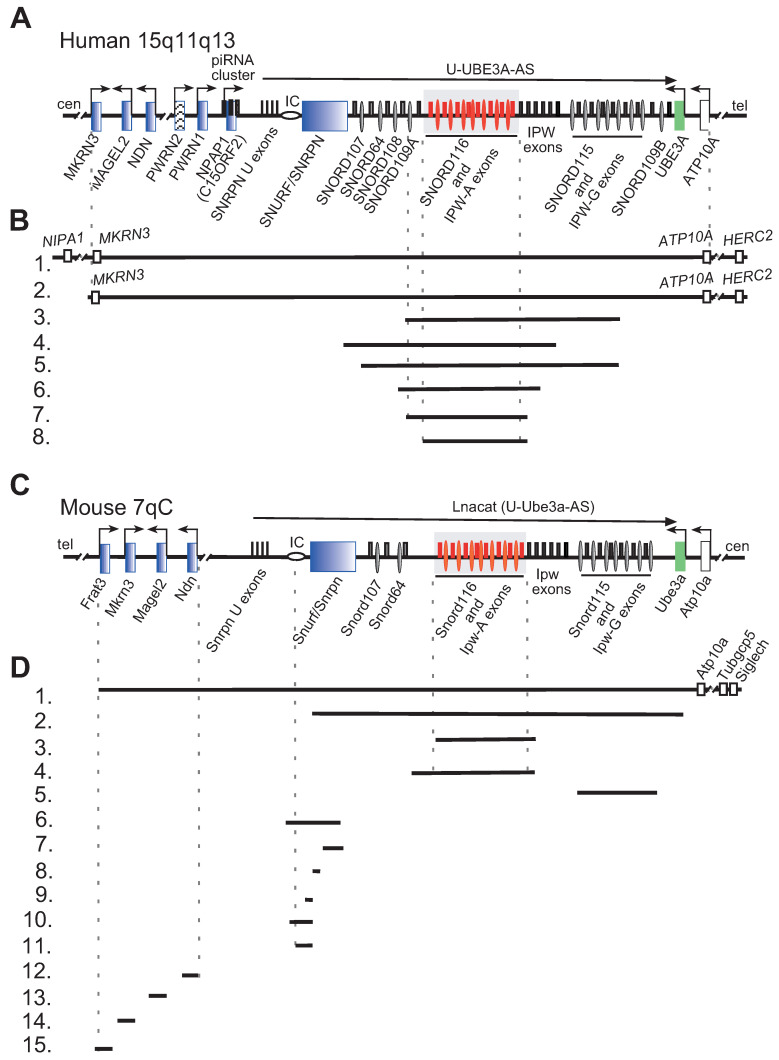
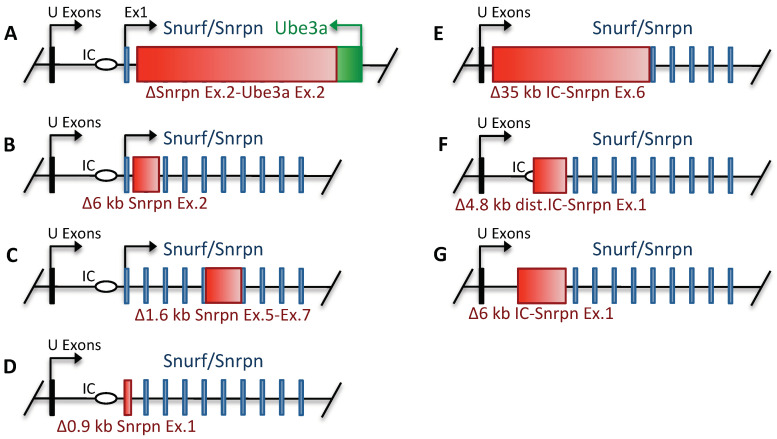
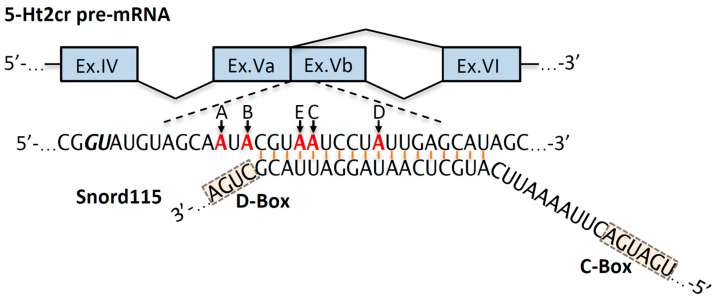
Prader-Willi syndrome (PWS) is a neurogenetic multifactorial disorder caused by the deletion or inactivation of paternally imprinted genes on human chromosome 15q11-q13. The affected homologous locus is on mouse chromosome 7C. The positional conservation and organization of genes including the imprinting pattern between mice and men implies similar physiological functions of this locus. Therefore, considerable efforts to recreate the pathogenesis of PWS have been accomplished in mouse models. We provide a summary of different mouse models that were generated for the analysis of PWS and discuss their impact on our current understanding of corresponding genes, their putative functions and the pathogenesis of PWS. Murine models of PWS unveiled the contribution of each affected gene to this multi-facetted disease, and also enabled the establishment of the minimal critical genomic region (PWScr) responsible for core symptoms, highlighting the importance of non-protein coding genes in the PWS locus. Although the underlying disease-causing mechanisms of PWS remain widely unresolved and existing mouse models do not fully capture the entire spectrum of the human PWS disorder, continuous improvements of genetically engineered mouse models have proven to be very powerful and valuable tools in PWS research.
Keywords: Magel2; PWS imprinting center (IC); Prader-Willi syndrome (PWS); Snord116; mouse models; non-coding RNAs.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
