

Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics
- PMID: 33807824
- PMCID: PMC8001122
- DOI: 10.3390/v13030417
Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics
Abstract
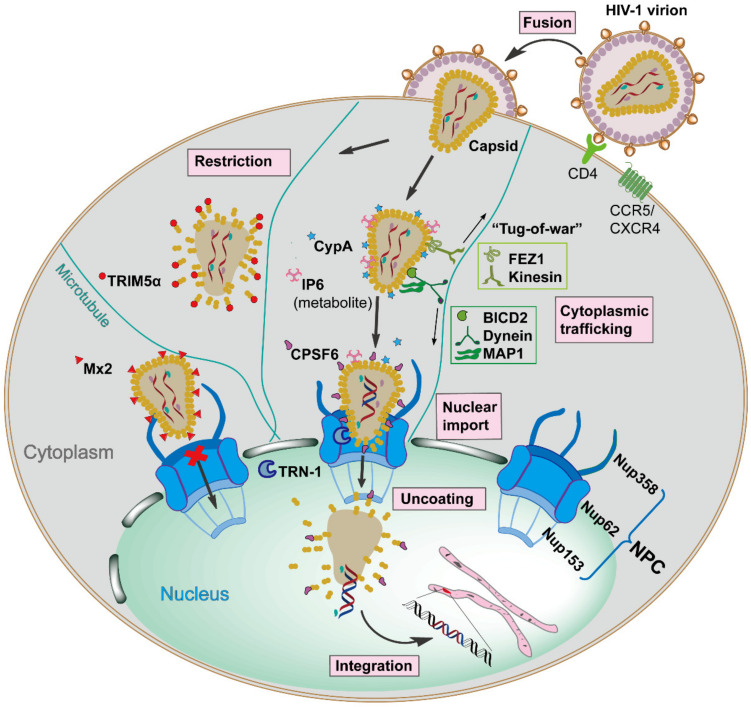
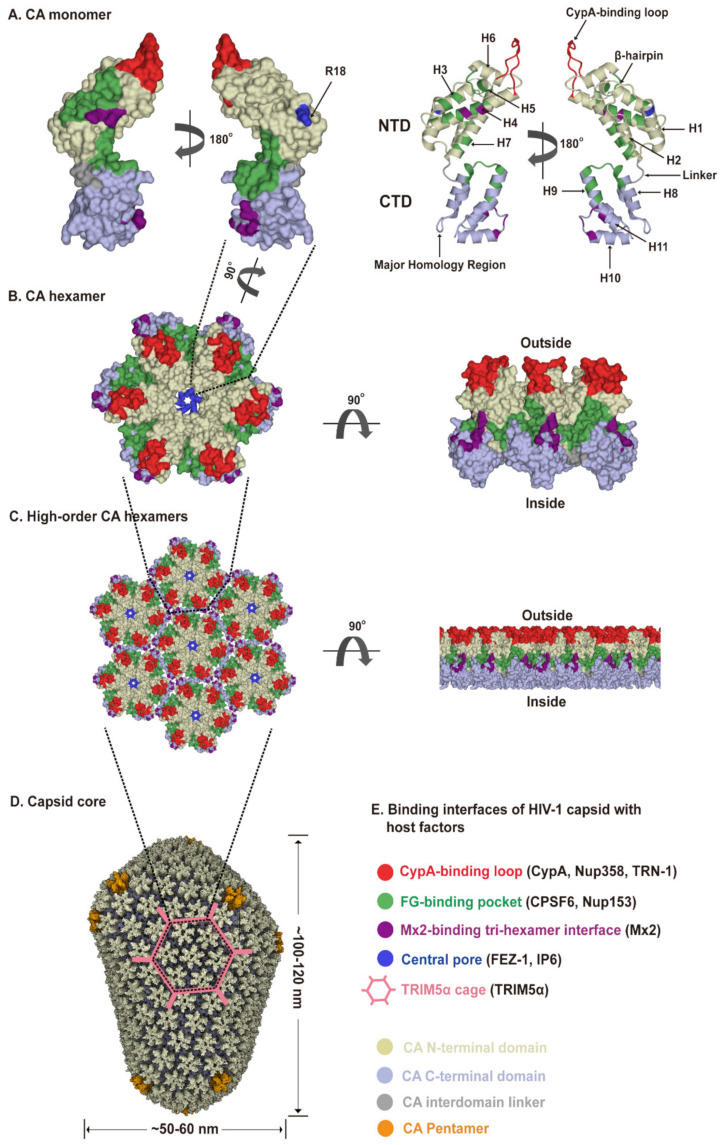
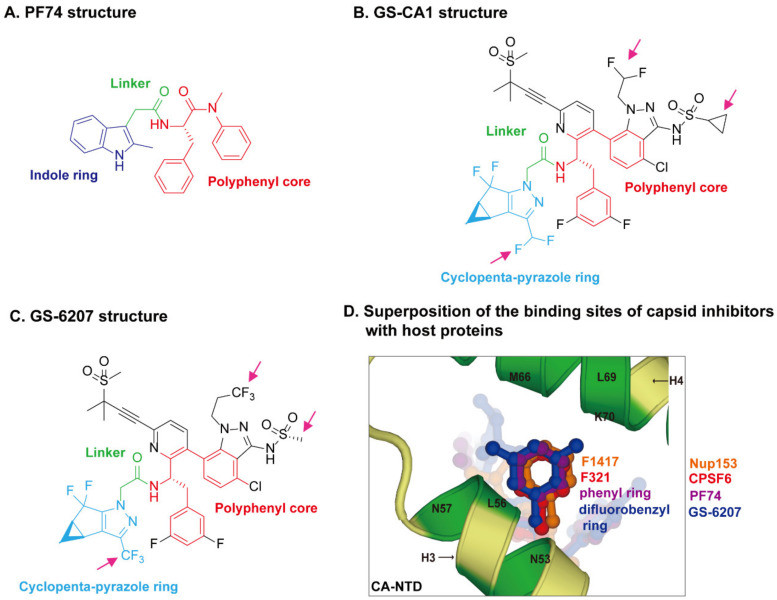
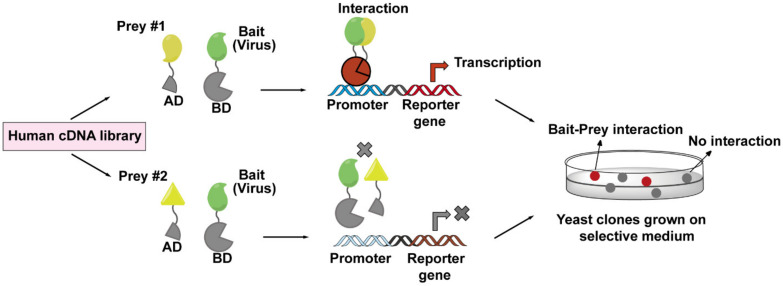
The Human Immunodeficiency Virus type 1 (HIV-1) virion contains a conical shell, termed capsid, encasing the viral RNA genome. After cellular entry of the virion, the capsid is released and ensures the protection and delivery of the HIV-1 genome to the host nucleus for integration. The capsid relies on many virus-host factor interactions which are regulated spatiotemporally throughout the course of infection. In this paper, we will review the current understanding of the highly dynamic HIV-1 capsid-host interplay during the early stages of viral replication, namely intracellular capsid trafficking after viral fusion, nuclear import, uncoating, and integration of the viral genome into host chromatin. Conventional anti-retroviral therapies primarily target HIV-1 enzymes. Insights of capsid structure have resulted in a first-in-class, long-acting capsid-targeting inhibitor, GS-6207 (Lenacapavir). This inhibitor binds at the interface between capsid protein subunits, a site known to bind host factors, interferes with capsid nuclear import, HIV particle assembly, and ordered assembly. Our review will highlight capsid structure, the host factors that interact with capsid, and high-throughput screening techniques, specifically genomic and proteomic approaches, that have been and can be used to identify host factors that interact with capsid. Better structural and mechanistic insights into the capsid-host factor interactions will significantly inform the understanding of HIV-1 pathogenesis and the development of capsid-centric antiretroviral therapeutics.
Keywords: HIV-1 capsid; antiretroviral therapeutics; capsid-targeting inhibitors; high-throughput screening techniques; host factors.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
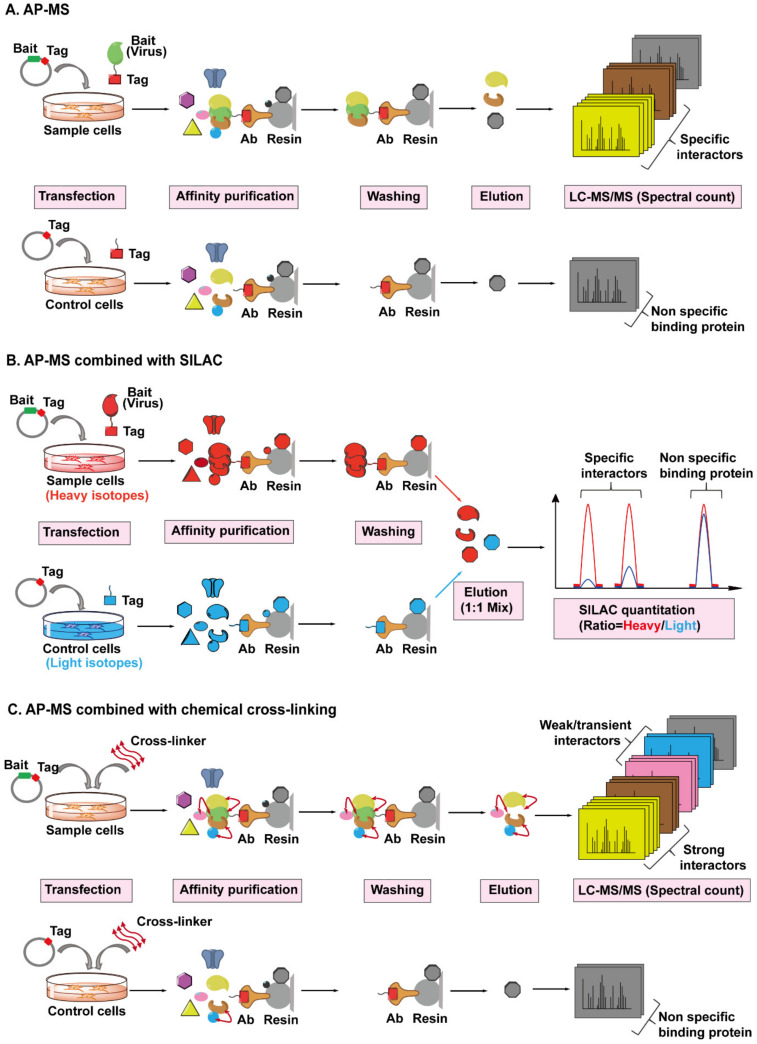
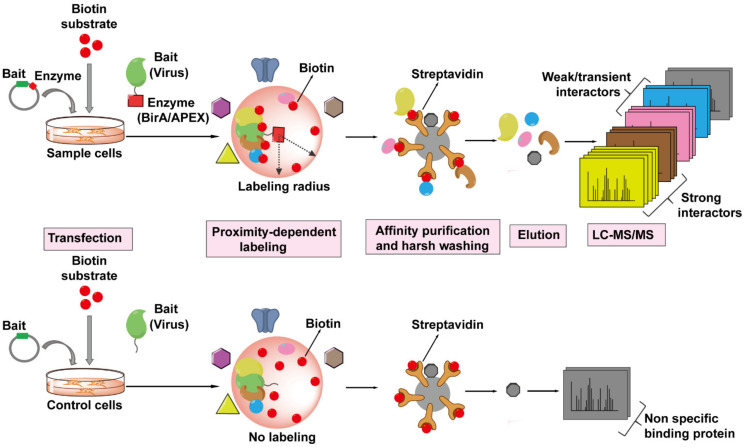
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
