

Metformin Attenuates Monosodium-Iodoacetate-Induced Osteoarthritis via Regulation of Pain Mediators and the Autophagy-Lysosomal Pathway
- PMID: 33808727
- PMCID: PMC8003384
- DOI: 10.3390/cells10030681
Metformin Attenuates Monosodium-Iodoacetate-Induced Osteoarthritis via Regulation of Pain Mediators and the Autophagy-Lysosomal Pathway
Abstract
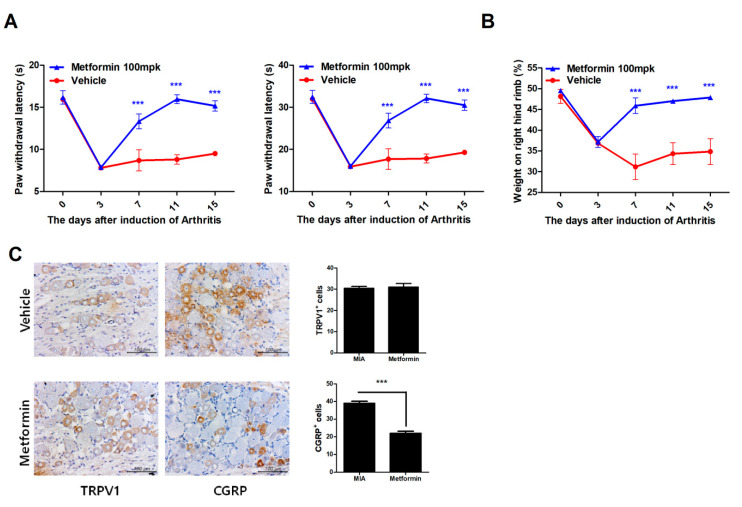
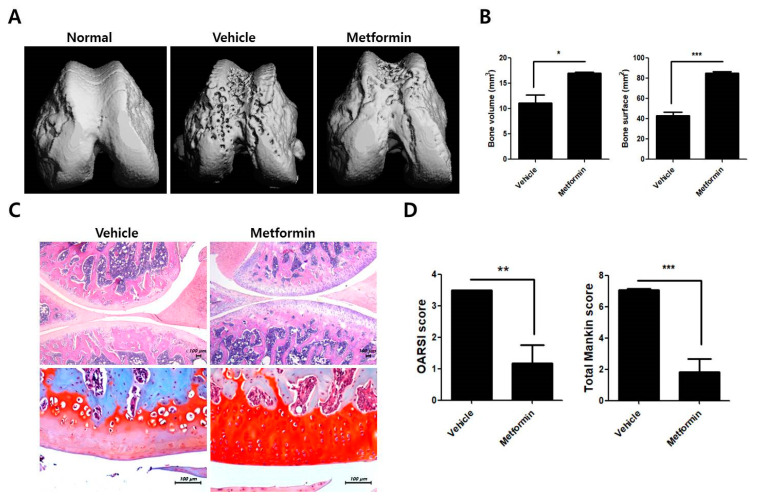
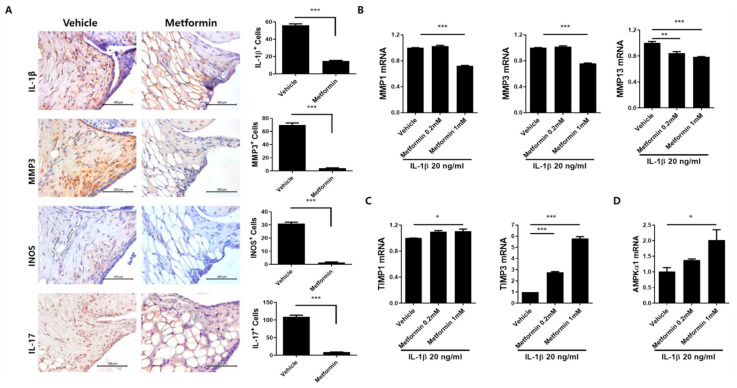
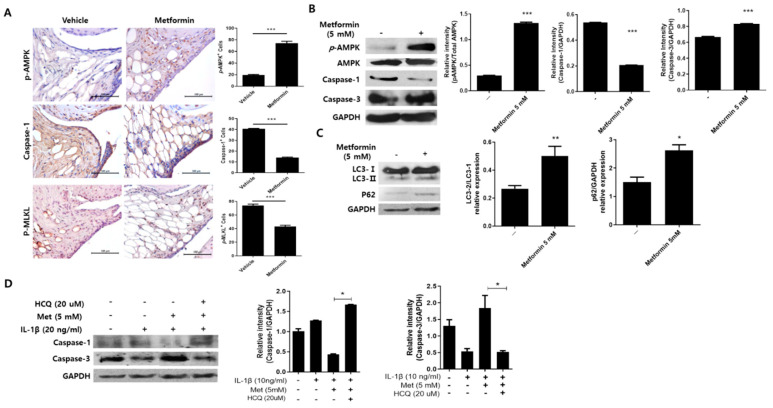
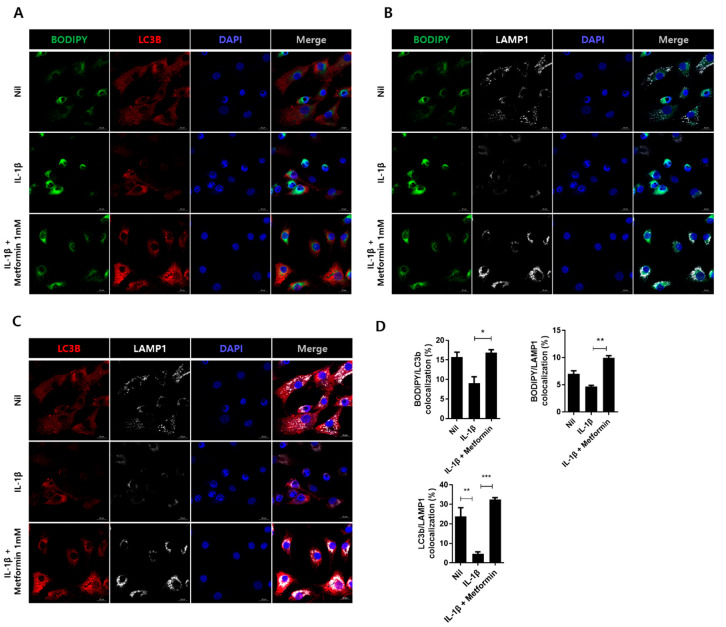
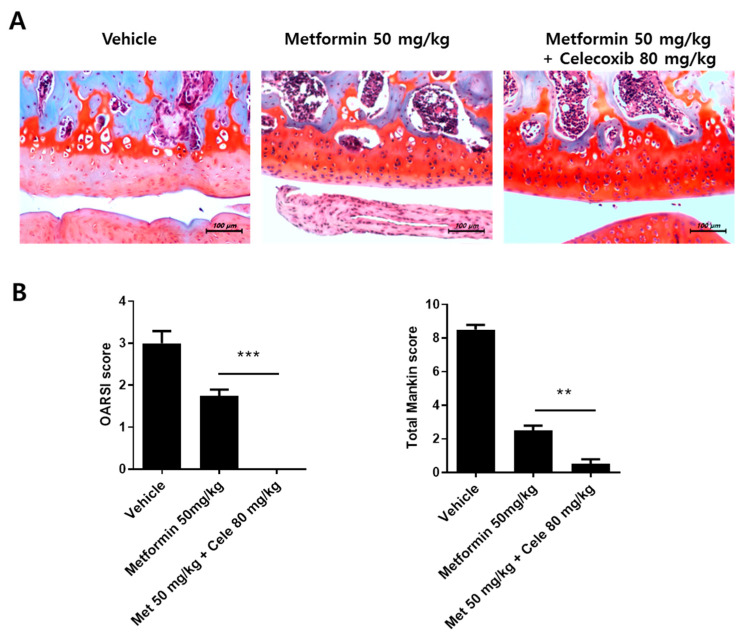
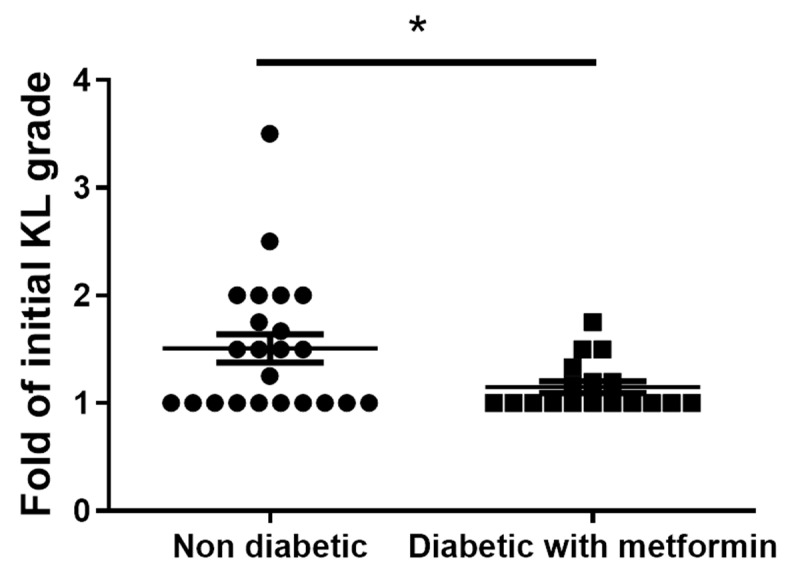
Osteoarthritis (OA) is the most common degenerative arthritis associated with pain and cartilage destruction in the elderly; it is known to be involved in inflammation as well. A drug called celecoxib is commonly used in patients with osteoarthritis to control pain. Metformin is used to treat type 2 diabetes but also exhibits regulation of the autophagy pathway. The purpose of this study is to investigate whether metformin can treat monosodium iodoacetate (MIA)-induced OA in rats. Metformin was administered orally every day to rats with OA. Paw-withdrawal latency and threshold were used to assess pain severity. Cartilage damage and pain mediators in dorsal root ganglia were evaluated by histological analysis and a scoring system. Relative mRNA expression was measured by real-time PCR. Metformin reduced the progression of experimental OA and showed both antinociceptive properties and cartilage protection. The combined administration of metformin and celecoxib controlled cartilage damage more effectively than metformin alone. In chondrocytes from OA patients, metformin reduced catabolic factor gene expression and inflammatory cell death factor expression, increased LC3Ⅱb, p62, and LAMP1 expression, and induced an autophagy-lysosome fusion phenotype. We investigated if metformin treatment reduces cartilage damage and inflammatory cell death of chondrocytes. The results suggest the potential for the therapeutic use of metformin in OA patients based on its ability to suppress pain and protect cartilage.
Keywords: autophagy; cartilage; combination therapy; metformin; osteoarthritis; pain.
Conflict of interest statement
The authors declare that they have no competing interest.
Figures
References
-
- Kalman D.S., Heimer M., Valdeon A., Schwartz H., Sheldon E. Effect of a natural extract of chicken combs with a high content of hyaluronic acid (Hyal-Joint) on pain relief and quality of life in subjects with knee osteoarthritis: A pilot randomized double-blind placebo-controlled trial. Nutr. J. 2008;7:3. doi: 10.1186/1475-2891-7-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
