

Mitochondria and Pharmacologic Cardiac Conditioning-At the Heart of Ischemic Injury
- PMID: 33810024
- PMCID: PMC8004818
- DOI: 10.3390/ijms22063224
Mitochondria and Pharmacologic Cardiac Conditioning-At the Heart of Ischemic Injury
Abstract
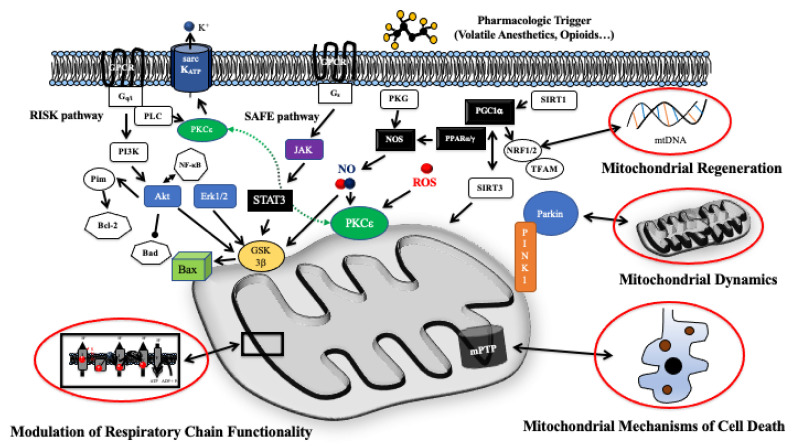
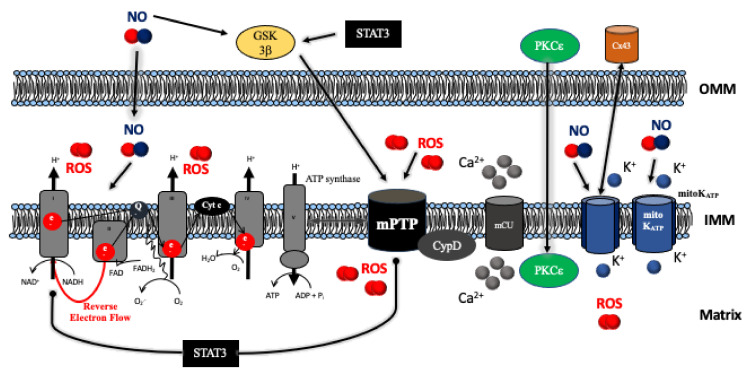
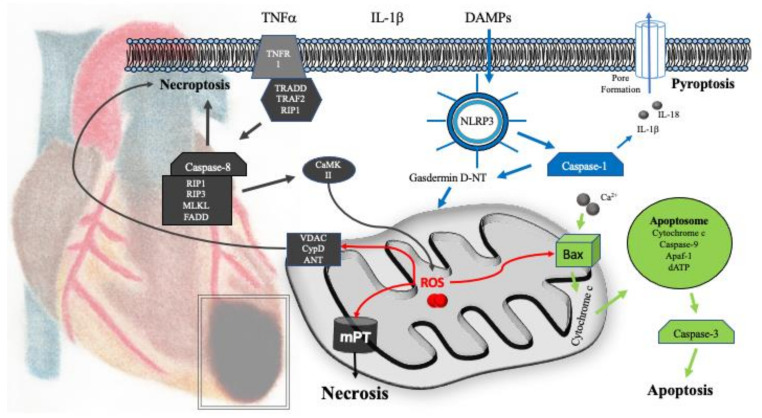
Pharmacologic cardiac conditioning increases the intrinsic resistance against ischemia and reperfusion (I/R) injury. The cardiac conditioning response is mediated via complex signaling networks. These networks have been an intriguing research field for decades, largely advancing our knowledge on cardiac signaling beyond the conditioning response. The centerpieces of this system are the mitochondria, a dynamic organelle, almost acting as a cell within the cell. Mitochondria comprise a plethora of functions at the crossroads of cell death or survival. These include the maintenance of aerobic ATP production and redox signaling, closely entwined with mitochondrial calcium handling and mitochondrial permeability transition. Moreover, mitochondria host pathways of programmed cell death impact the inflammatory response and contain their own mechanisms of fusion and fission (division). These act as quality control mechanisms in cellular ageing, release of pro-apoptotic factors and mitophagy. Furthermore, recently identified mechanisms of mitochondrial regeneration can increase the capacity for oxidative phosphorylation, decrease oxidative stress and might help to beneficially impact myocardial remodeling, as well as invigorate the heart against subsequent ischemic insults. The current review highlights different pathways and unresolved questions surrounding mitochondria in myocardial I/R injury and pharmacological cardiac conditioning.
Keywords: cardioprotection; ischemia/reperfusion injury; preconditioning; volatile anesthetics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Albrecht M., Zitta K., Bein B., Wennemuth G., Broch O., Renner J., Schuett T., Lauer F., Maahs D., Hummitzsch L., et al. Remote ischemic preconditioning regulates HIF-1alpha levels, apoptosis and inflammation in heart tissue of cardiosurgical patients: A pilot experimental study. Basic Res. Cardiol. 2013;108:314. doi: 10.1007/s00395-012-0314-0. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
