

Metastasis-Initiating Cells and Ecosystems
- PMID: 33811127
- PMCID: PMC8030695
- DOI: 10.1158/2159-8290.CD-21-0010
Metastasis-Initiating Cells and Ecosystems
Abstract
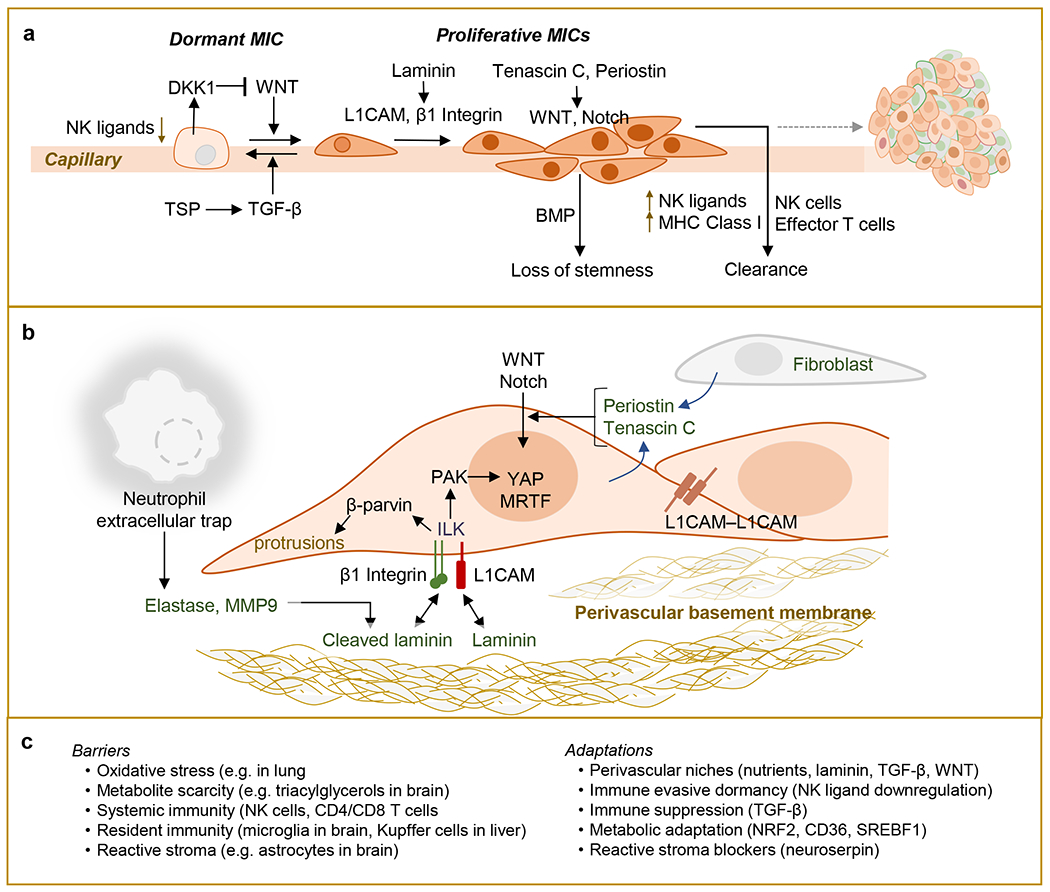
Metastasis is initiated and sustained through therapy by cancer cells with stem-like and immune-evasive properties, termed metastasis-initiating cells (MIC). Recent progress suggests that MICs result from the adoption of a normal regenerative progenitor phenotype by malignant cells, a phenotype with intrinsic programs to survive the stresses of the metastatic process, undergo epithelial-mesenchymal transitions, enter slow-cycling states for dormancy, evade immune surveillance, establish supportive interactions with organ-specific niches, and co-opt systemic factors for growth and recurrence after therapy. Mechanistic understanding of the molecular mediators of MIC phenotypes and host tissue ecosystems could yield cancer therapeutics to improve patient outcomes. SIGNIFICANCE: Understanding the origins, traits, and vulnerabilities of progenitor cancer cells with the capacity to initiate metastasis in distant organs, and the host microenvironments that support the ability of these cells to evade immune surveillance and regenerate the tumor, is critical for developing strategies to improve the prevention and treatment of advanced cancer. Leveraging recent progress in our understanding of the metastatic process, here we review the nature of MICs and their ecosystems and offer a perspective on how this knowledge is informing innovative treatments of metastatic cancers.
©2021 American Association for Cancer Research.
Conflict of interest statement
Figures





References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
