

Structural and Biochemical Analysis of OrfG: The VirB8-like Component of the Conjugative Type IV Secretion System of ICE St3 From Streptococcus thermophilus
- PMID: 33816557
- PMCID: PMC8012802
- DOI: 10.3389/fmolb.2021.642606
Structural and Biochemical Analysis of OrfG: The VirB8-like Component of the Conjugative Type IV Secretion System of ICE St3 From Streptococcus thermophilus
Abstract
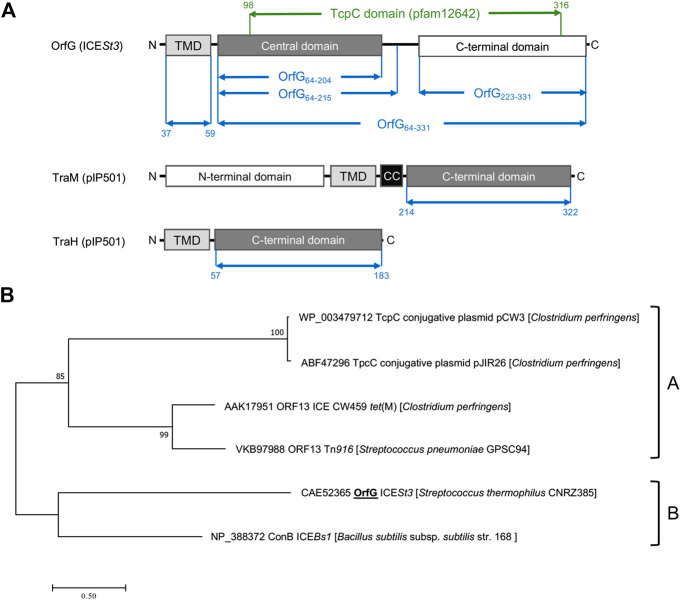
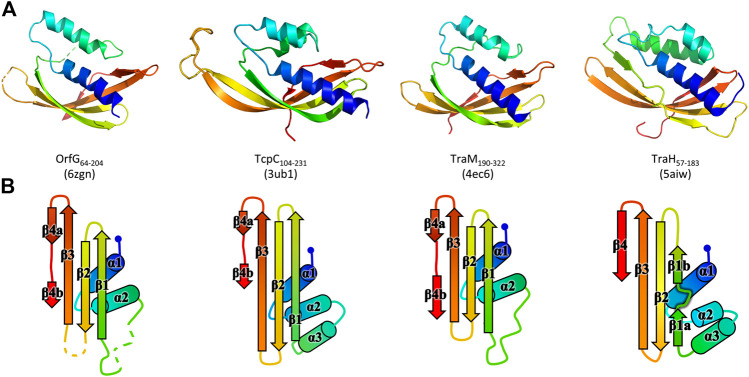
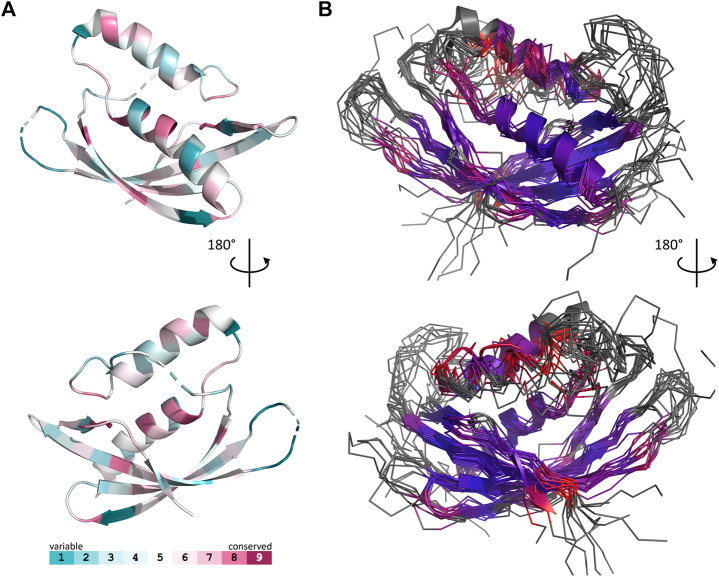
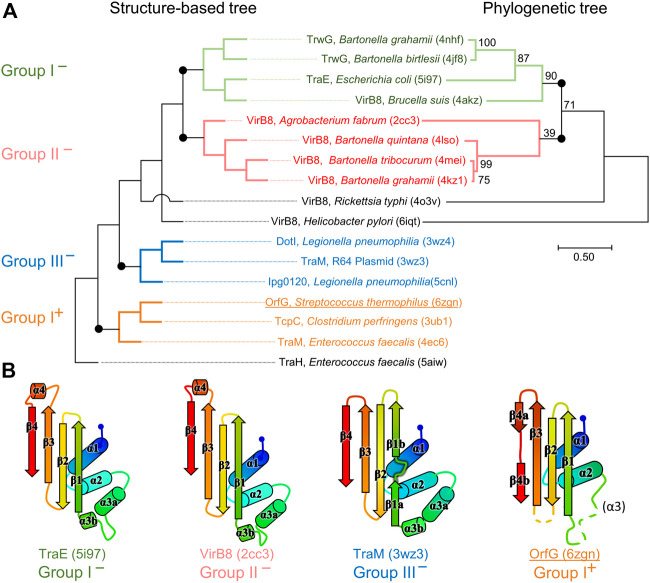
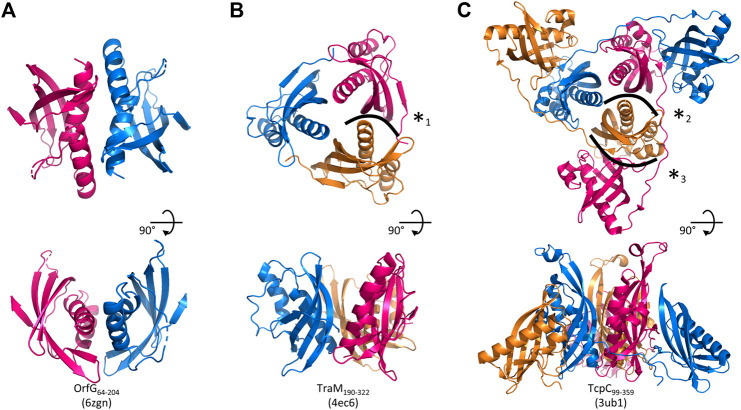
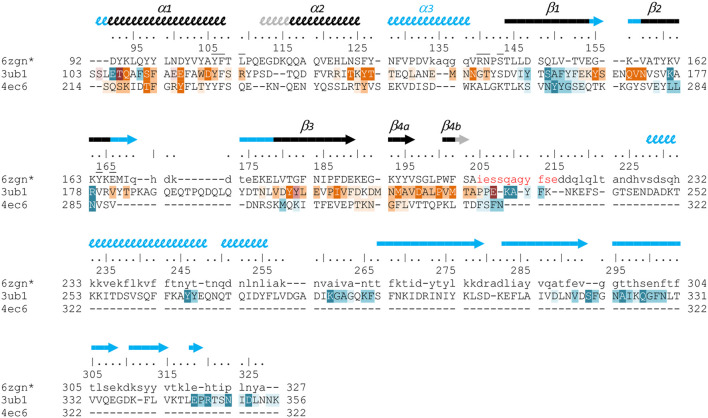
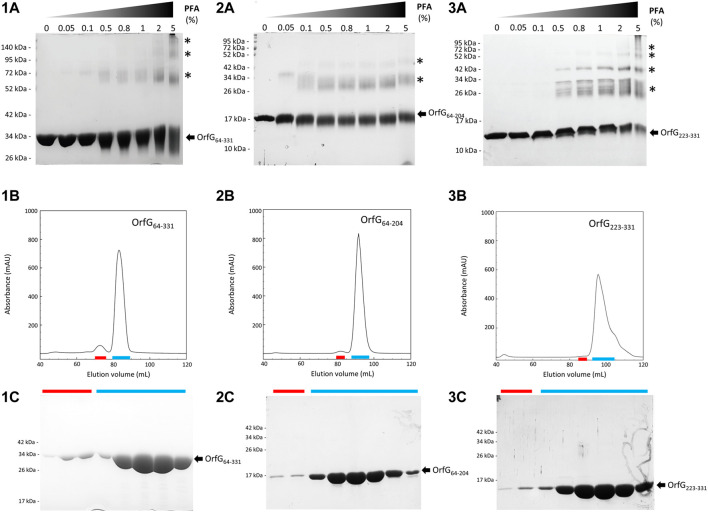
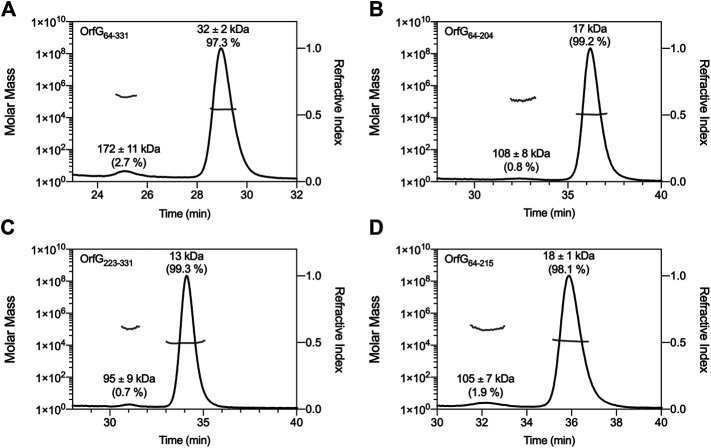
Conjugative transfer is a major threat to global health since it contributes to the spread of antibiotic resistance genes and virulence factors among commensal and pathogenic bacteria. To allow their transfer, mobile genetic elements including Integrative and Conjugative Elements (ICEs) use a specialized conjugative apparatus related to Type IV secretion systems (Conj-T4SS). Therefore, Conj-T4SSs are excellent targets for strategies that aim to limit the spread of antibiotic resistance. In this study, we combined structural, biochemical and biophysical approaches to study OrfG, a protein that belongs to Conj-T4SS of ICESt3 from Streptococcus thermophilus. Structural analysis of OrfG by X-ray crystallography revealed that OrfG central domain is similar to VirB8-like proteins but displays a different quaternary structure in the crystal. To understand, at a structural level, the common and the diverse features between VirB8-like proteins from both Gram-negative and -positive bacteria, we used an in silico structural alignment method that allowed us to identify different structural classes of VirB8-like proteins. Biochemical and biophysical characterizations of purified OrfG soluble domain and its central and C-terminal subdomains indicated that they are mainly monomeric in solution but able to form an unprecedented 6-mer oligomers. Our study provides new insights into the structural analysis of VirB8-like proteins and discusses the interplay between tertiary and quaternary structures of these proteins as an essential component of the conjugative transfer.
Keywords: Gram - positive bacteria; Integrative and Conjugative Element (ICE); VirB8-like proteins; conjugation; type IV secretion system.
Copyright © 2021 Cappele, Mohamad Ali, Leblond-Bourget, Mathiot, Dhalleine, Payot, Savko, Didierjean, Favier and Douzi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Diversity of Integrative and Conjugative Elements of Streptococcus salivarius and Their Intra- and Interspecies Transfer.Appl Environ Microbiol. 2017 Jun 16;83(13):e00337-17. doi: 10.1128/AEM.00337-17. Print 2017 Jul 1. Appl Environ Microbiol. 2017. PMID: 28432093 Free PMC article.
-
Structural Insight into How Bacteria Prevent Interference between Multiple Divergent Type IV Secretion Systems.mBio. 2015 Dec 8;6(6):e01867-15. doi: 10.1128/mBio.01867-15. mBio. 2015. PMID: 26646013 Free PMC article.
-
Impact of Cell Surface Molecules on Conjugative Transfer of the Integrative and Conjugative Element ICESt3 of Streptococcus thermophilus.Appl Environ Microbiol. 2018 Feb 14;84(5):e02109-17. doi: 10.1128/AEM.02109-17. Print 2018 Mar 1. Appl Environ Microbiol. 2018. PMID: 29247061 Free PMC article.
-
Regulation of Gram-Positive Conjugation.Front Microbiol. 2019 May 22;10:1134. doi: 10.3389/fmicb.2019.01134. eCollection 2019. Front Microbiol. 2019. PMID: 31191478 Free PMC article. Review.
-
Advances in Protein Structure Prediction Highlight Unexpected Commonalities Between Gram-positive and Gram-negative Conjugative T4SSs.J Mol Biol. 2025 Feb 15;437(4):168924. doi: 10.1016/j.jmb.2024.168924. Epub 2024 Dec 31. J Mol Biol. 2025. PMID: 39746464 Review.
Cited by
-
Transcriptional control of two distinct lactococcal plasmid-encoded conjugation systems.Curr Res Microb Sci. 2024 Feb 5;6:100224. doi: 10.1016/j.crmicr.2024.100224. eCollection 2024. Curr Res Microb Sci. 2024. PMID: 38371911 Free PMC article.
-
Elucidating assembly and function of VirB8 cell wall subunits refines the DNA translocation model in Gram-positive T4SSs.Sci Adv. 2025 Jan 24;11(4):eadq5975. doi: 10.1126/sciadv.adq5975. Epub 2025 Jan 22. Sci Adv. 2025. PMID: 39841841 Free PMC article.
References
-
- Ambroset C., Coluzzi C., Guédon G., Devignes M. D., Loux V., Lacroix T., et al. (2015). New insights into the classification and integration specificity of Streptococcus integrative conjugative elements through extensive genome exploration. Front. Microbiol. 6, 1483. 10.3389/fmicb.2015.01483 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous