

A machine learning framework for the prediction of chromatin folding in Drosophila using epigenetic features
- PMID: 33816958
- PMCID: PMC7924456
- DOI: 10.7717/peerj-cs.307
A machine learning framework for the prediction of chromatin folding in Drosophila using epigenetic features
Abstract
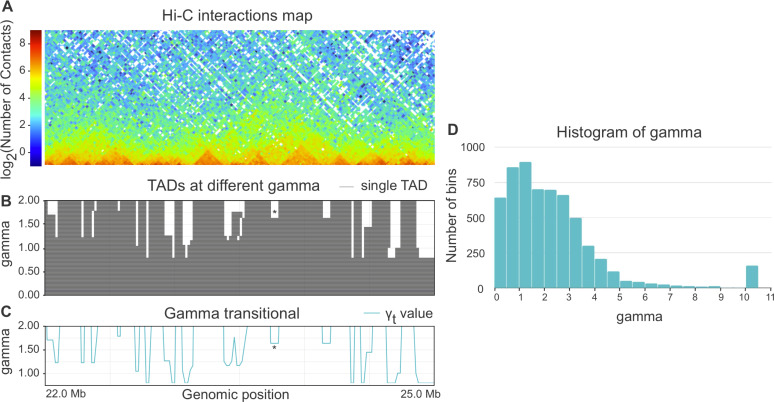
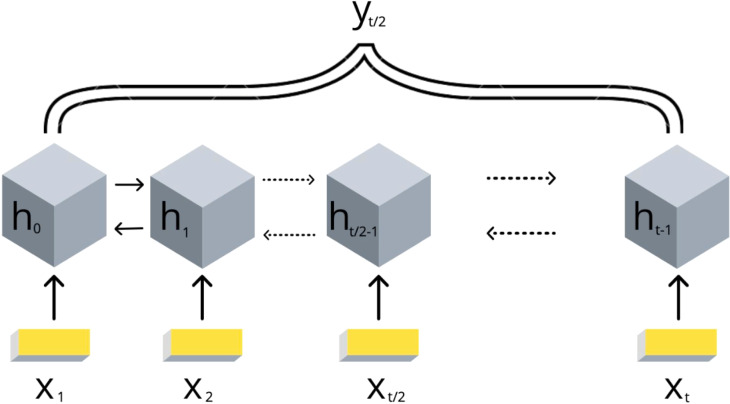
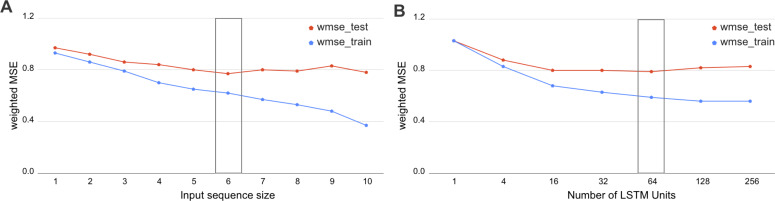
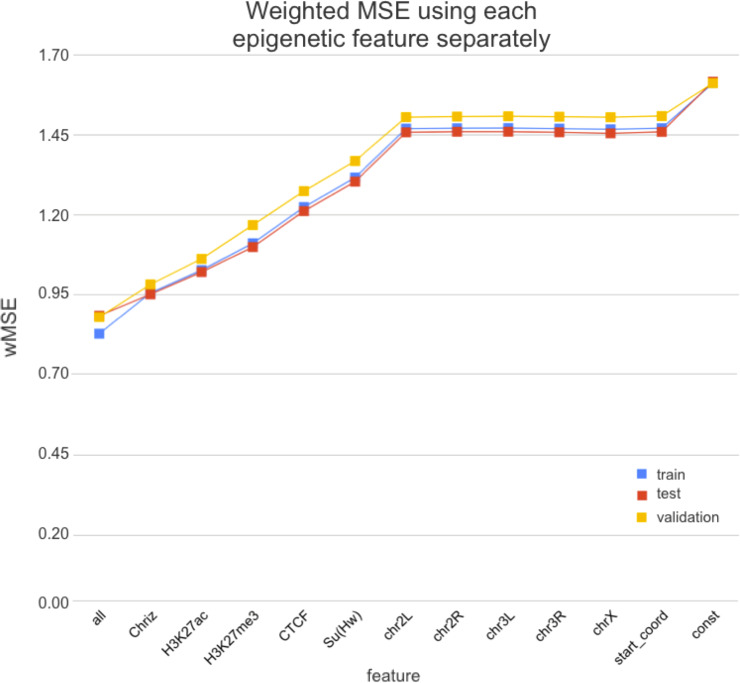
Technological advances have lead to the creation of large epigenetic datasets, including information about DNA binding proteins and DNA spatial structure. Hi-C experiments have revealed that chromosomes are subdivided into sets of self-interacting domains called Topologically Associating Domains (TADs). TADs are involved in the regulation of gene expression activity, but the mechanisms of their formation are not yet fully understood. Here, we focus on machine learning methods to characterize DNA folding patterns in Drosophila based on chromatin marks across three cell lines. We present linear regression models with four types of regularization, gradient boosting, and recurrent neural networks (RNN) as tools to study chromatin folding characteristics associated with TADs given epigenetic chromatin immunoprecipitation data. The bidirectional long short-term memory RNN architecture produced the best prediction scores and identified biologically relevant features. Distribution of protein Chriz (Chromator) and histone modification H3K4me3 were selected as the most informative features for the prediction of TADs characteristics. This approach may be adapted to any similar biological dataset of chromatin features across various cell lines and species. The code for the implemented pipeline, Hi-ChiP-ML, is publicly available: https://github.com/MichalRozenwald/Hi-ChIP-ML.
Keywords: Chromatin; DNA folding patterns; Gradient Boosting; Hi-C experiments; Linear Regression; Machine Learning; Recurrent Neural Networks (RNN); Topologically Associating Domains (TADs).
©2020 Rozenwald et al.
Conflict of interest statement
Mikhail Gelfand is an Academic Editor for PeerJ. Grigory V. Sapunov is employed by Intento, Inc.
Figures
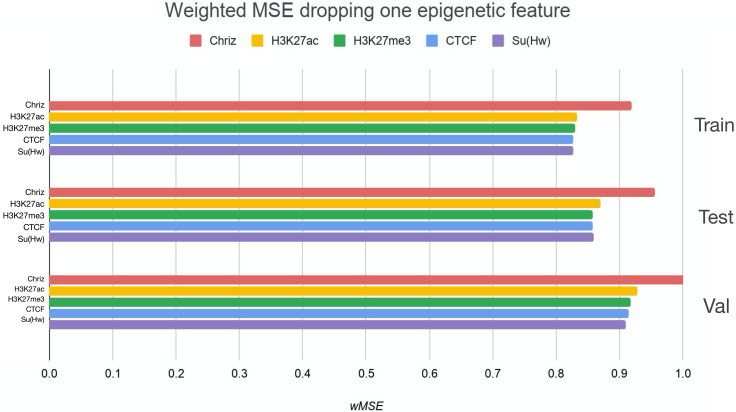
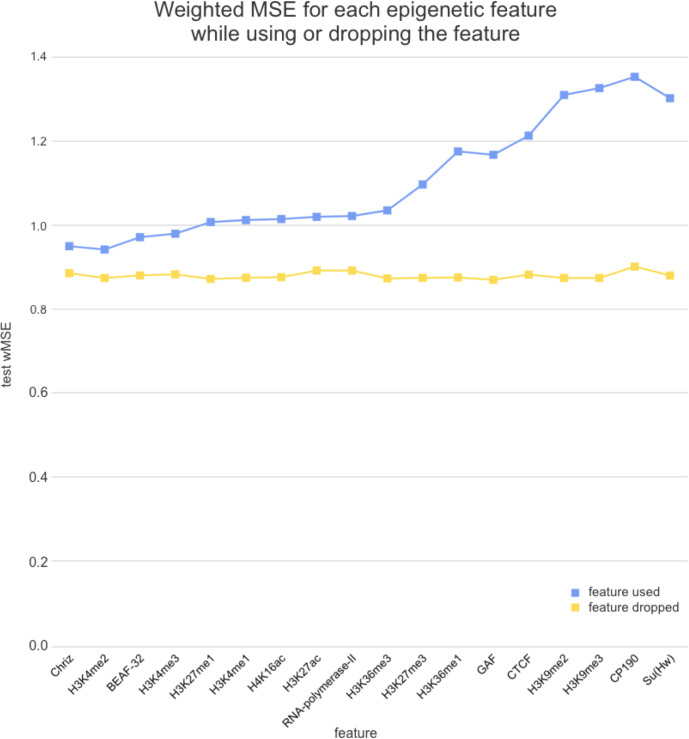
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases