

Pharmacological and nutritional targeting of voltage-gated sodium channels in the treatment of cancers
- PMID: 33817575
- PMCID: PMC8010468
- DOI: 10.1016/j.isci.2021.102270
Pharmacological and nutritional targeting of voltage-gated sodium channels in the treatment of cancers
Abstract
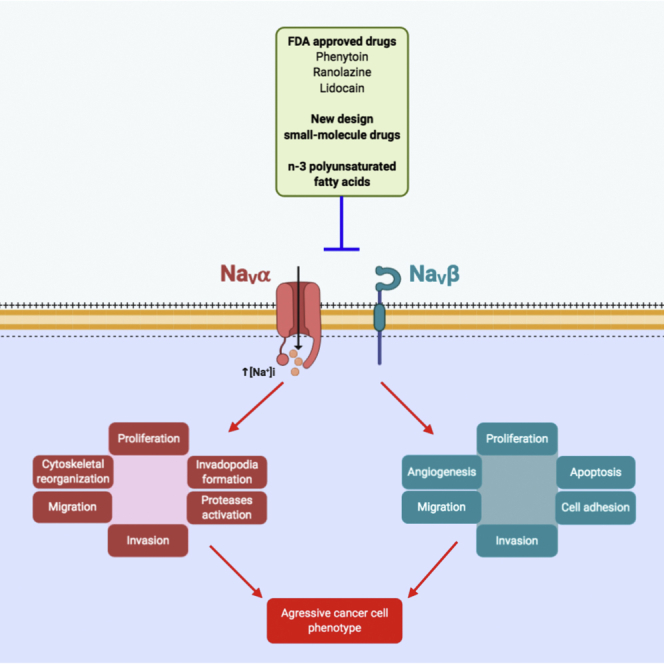
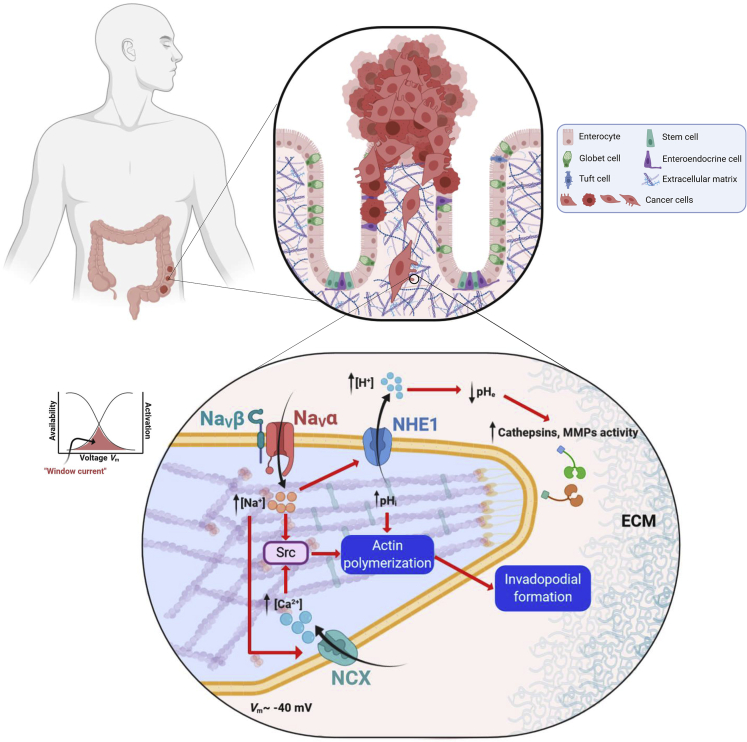
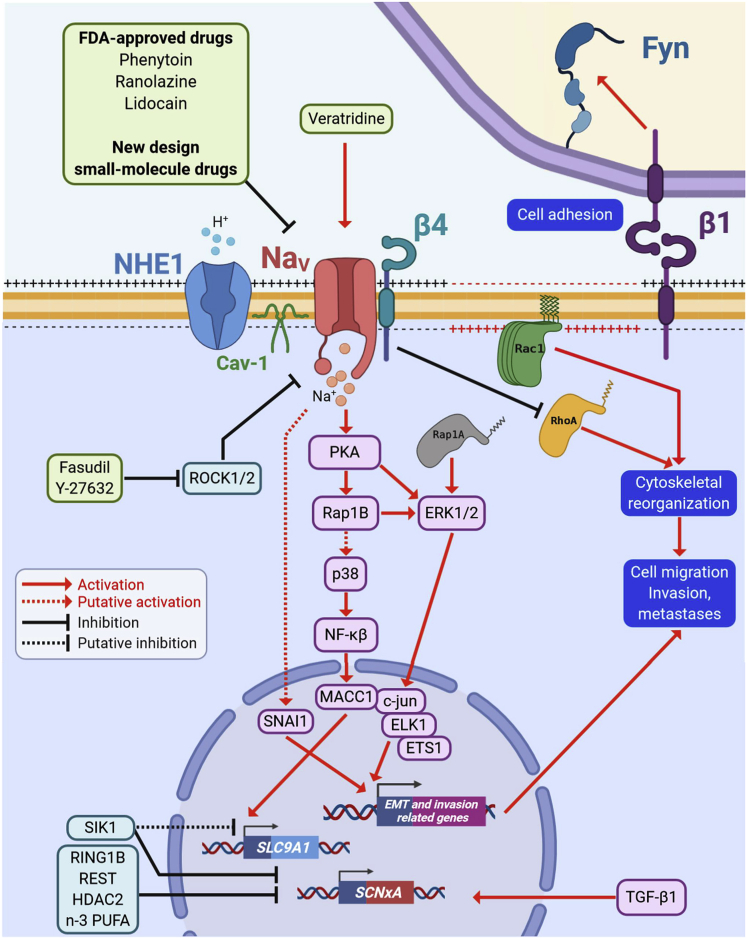
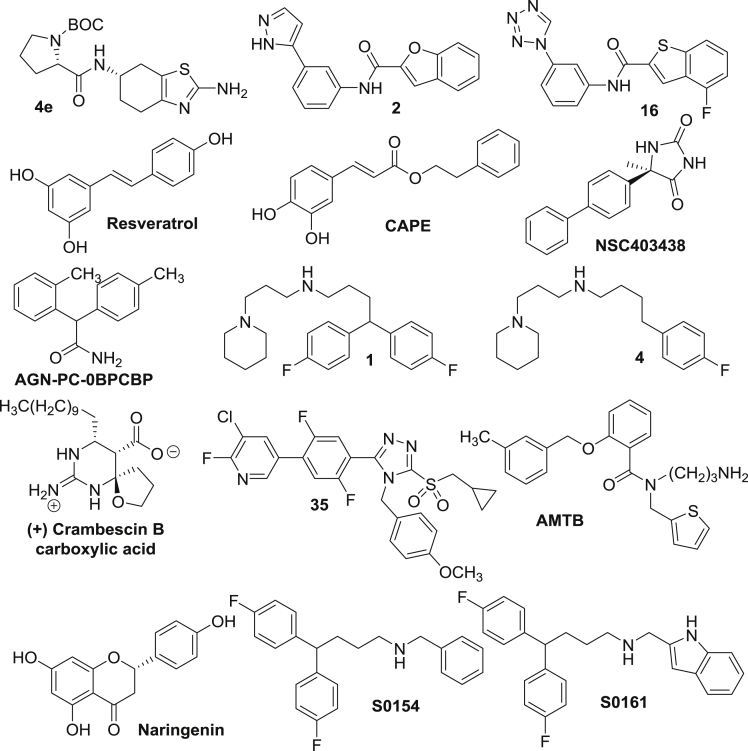
Voltage-gated sodium (NaV) channels, initially characterized in excitable cells, have been shown to be aberrantly expressed in non-excitable cancer tissues and cells from epithelial origins such as in breast, lung, prostate, colon, and cervix, whereas they are not expressed in cognate non-cancer tissues. Their activity was demonstrated to promote aggressive and invasive potencies of cancer cells, both in vitro and in vivo, whereas their deregulated expression in cancer tissues has been associated with metastatic progression and cancer-related death. This review proposes NaV channels as pharmacological targets for anticancer treatments providing opportunities for repurposing existing NaV-inhibitors or developing new pharmacological and nutritional interventions.
Keywords: Cancer; Cell Biology.
© 2021 The Author(s).
Conflict of interest statement
Authors declare no competing interest.
Figures
References
-
- Adachi K., Toyota M., Sasaki Y., Yamashita T., Ishida S., Ohe-Toyota M., Maruyama R., Hinoda Y., Saito T., Imai K. Identification of SCN3B as a novel p53-inducible proapoptotic gene. Oncogene. 2004;23:7791–7798. - PubMed
-
- Agnew W.S., Moore A.C., Levinson S.R., Raftery M.A. Identification of a large molecular weight peptide associated with a tetrodotoxin binding protein from the electroplax of Electrophorus electricus. Biochem. Biophys. Res. Commun. 1980;92:860–866. - PubMed
-
- Arnold M., Sierra M.S., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–691. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
