

Mutations in phospholipase C eta-1 (PLCH1) are associated with holoprosencephaly
- PMID: 33820834
- PMCID: PMC8961749
- DOI: 10.1136/jmedgenet-2020-107237
Mutations in phospholipase C eta-1 (PLCH1) are associated with holoprosencephaly
Abstract
Background: Holoprosencephaly is a spectrum of developmental disorder of the embryonic forebrain in which there is failed or incomplete separation of the prosencephalon into two cerebral hemispheres. To date, dominant mutations in sonic hedgehog (SHH) pathway genes are the predominant Mendelian causes, and have marked interfamilial and intrafamilial phenotypical variabilities.
Methods: We describe two families in which offspring had holoprosencephaly spectrum and homozygous predicted-deleterious variants in phospholipase C eta-1 (PLCH1). Immunocytochemistry was used to examine the expression pattern of PLCH1 in human embryos. We used SHH as a marker of developmental stage and of early embryonic anatomy.
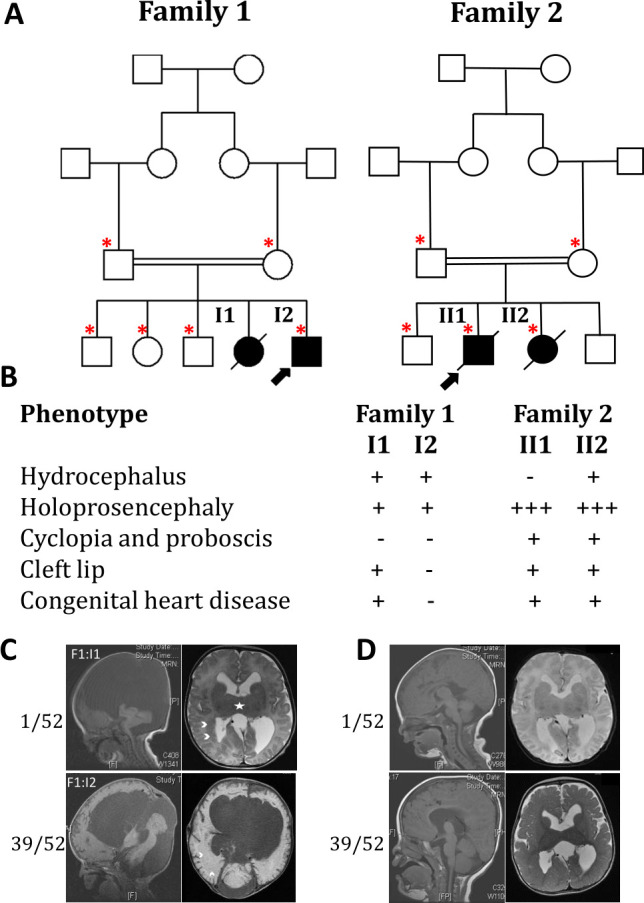
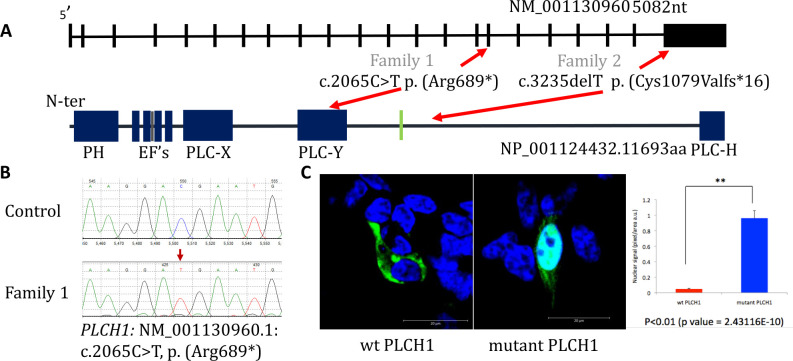
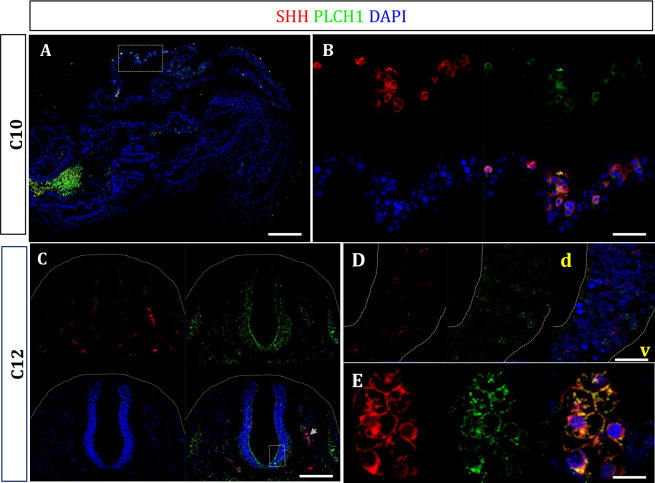
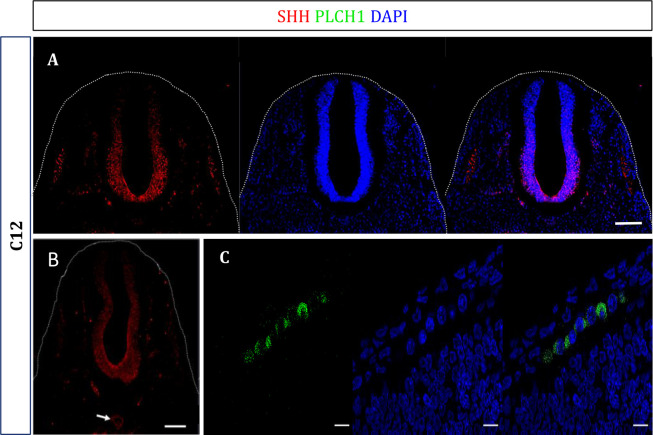
Results: In the first family, two siblings had congenital hydrocephalus, significant developmental delay and a monoventricle or fused thalami with a homozygous PLCH1 c.2065C>T, p.(Arg689*) variant. In the second family, two siblings had alobar holoprosencephaly and cyclopia with a homozygous PLCH1 c.4235delA, p.(Cys1079ValfsTer16) variant. All parents were healthy carriers, with no holoprosencephaly spectrum features. We found that the subcellular localisation of PLCH1 is cytoplasmic, but the p.(Cys1079ValfsTer16) variant was predominantly nuclear. Human embryo immunohistochemistry showed PLCH1 to be expressed in the notorcord, developing spinal cord (in a ventral to dorsal gradient), dorsal root ganglia, cerebellum and dermatomyosome, all tissues producing or responding to SHH. Furthermore, the embryonic subcellular localisation of PLCH1 was exclusively cytoplasmic, supporting protein mislocalisation contributing to the pathogenicity of the p.(Cys1079ValfsTer16) variant.
Conclusion: Our data support the contention that PLCH1 has a role in prenatal mammalian neurodevelopment, and deleterious variants cause a clinically variable holoprosencephaly spectrum phenotype.
Keywords: and neonatal diseases and abnormalities; cerebellar diseases; congenital; genetics; hereditary; mutation.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
Similar articles
-
Alobar Holoprosencephaly in an Aborted American Quarter Horse Fetus.J Equine Vet Sci. 2022 May;112:103898. doi: 10.1016/j.jevs.2022.103898. Epub 2022 Feb 9. J Equine Vet Sci. 2022. PMID: 35150851
-
Wide phenotypic variability in families with holoprosencephaly and a sonic hedgehog mutation.Eur J Pediatr. 2004 Jul;163(7):347-52. doi: 10.1007/s00431-004-1459-0. Epub 2004 Apr 24. Eur J Pediatr. 2004. PMID: 15107988
-
The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly.Hum Mol Genet. 1999 Dec;8(13):2479-88. doi: 10.1093/hmg/8.13.2479. Hum Mol Genet. 1999. PMID: 10556296
-
Patterning of the antero-ventral mammalian brain: Lessons from holoprosencephaly comparative biology in man and mouse.WIREs Mech Dis. 2022 Jul;14(4):e1552. doi: 10.1002/wsbm.1552. Epub 2022 Feb 8. WIREs Mech Dis. 2022. PMID: 35137563 Review.
-
Mouse models of holoprosencephaly.Curr Opin Neurol. 2003 Apr;16(2):135-41. doi: 10.1097/01.wco.0000063761.15877.40. Curr Opin Neurol. 2003. PMID: 12644739 Review.
Cited by
-
Cyclopia in a newborn rhesus macaque born to a dam infected with SIV and receiving antiretroviral therapy during pregnancy.Curr Trends Immunol. 2023;24:91-103. Curr Trends Immunol. 2023. PMID: 39640529 Free PMC article.
-
Phosphoinositide Metabolism: Biochemistry, Physiology and Genetic Disorders.J Inherit Metab Dis. 2025 Mar;48(2):e70008. doi: 10.1002/jimd.70008. J Inherit Metab Dis. 2025. PMID: 40024625 Free PMC article. Review.
-
Concepts in Multifactorial Etiology of Developmental Disorders: Gene-Gene and Gene-Environment Interactions in Holoprosencephaly.Front Cell Dev Biol. 2021 Dec 22;9:795194. doi: 10.3389/fcell.2021.795194. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 35004690 Free PMC article. Review.
References
-
- Solomon BD, Gropman A, Muenke M. Holoprosencephaly Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, 2000: 1993–2019.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous