

Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases
- PMID: 33823185
- PMCID: PMC8018918
- DOI: 10.1016/j.ejphar.2021.174082
Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases
Abstract
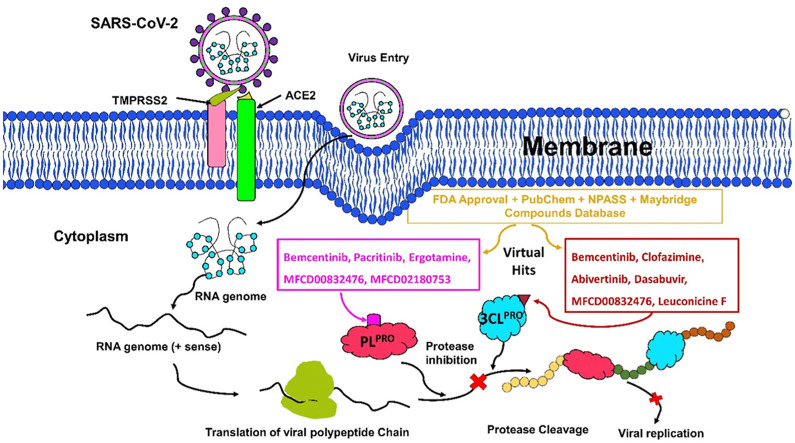
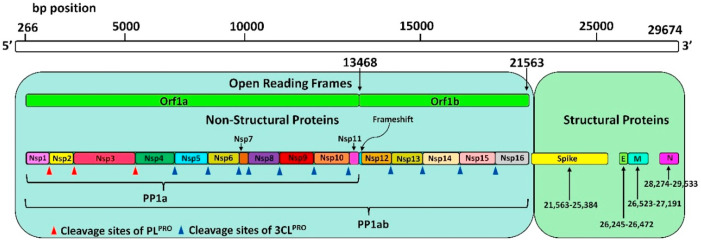
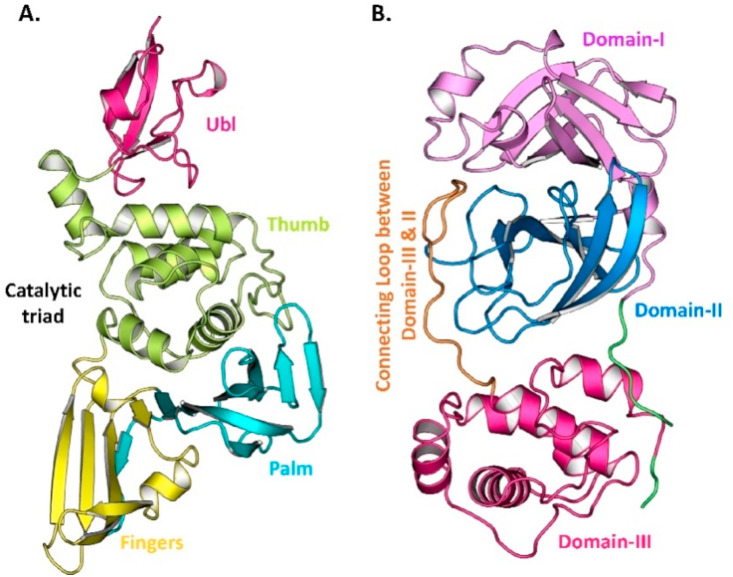
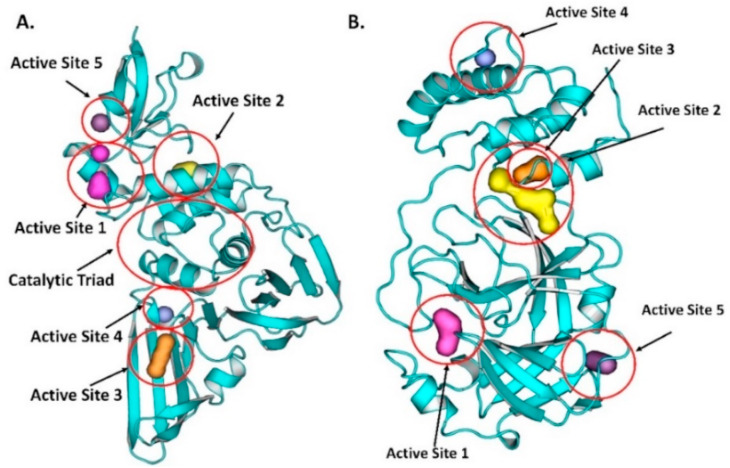
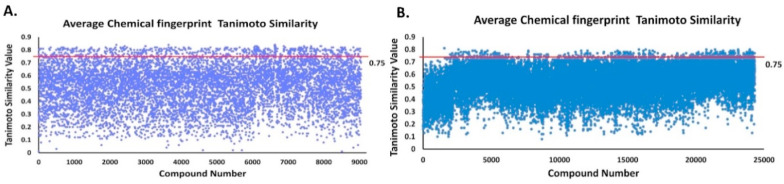
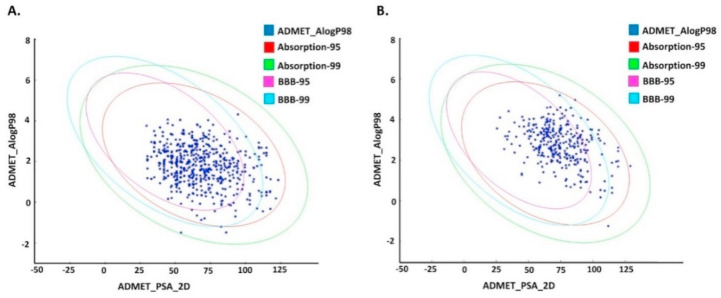
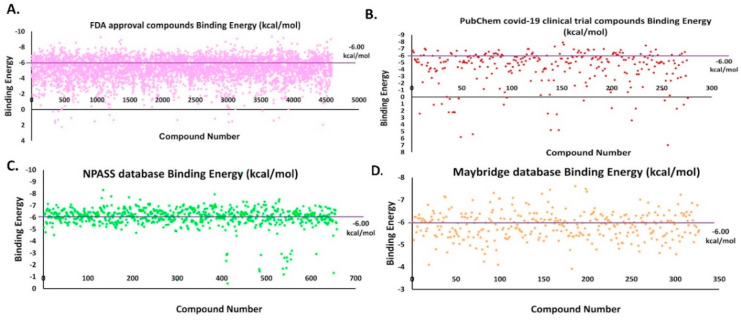
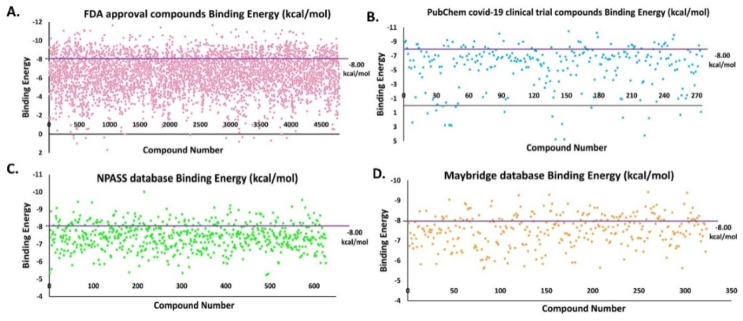
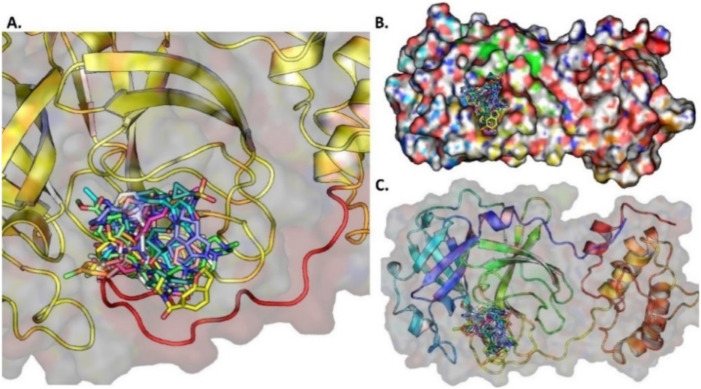
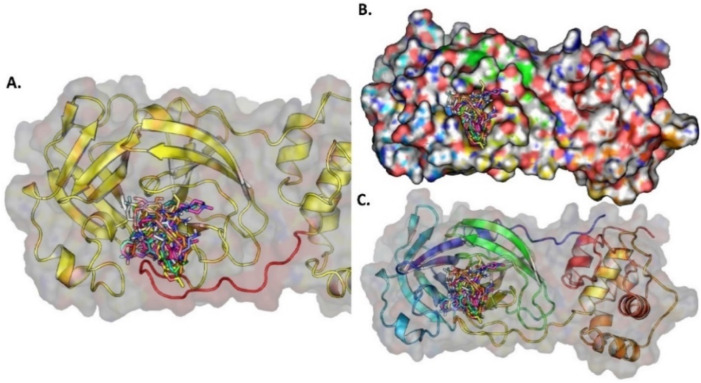
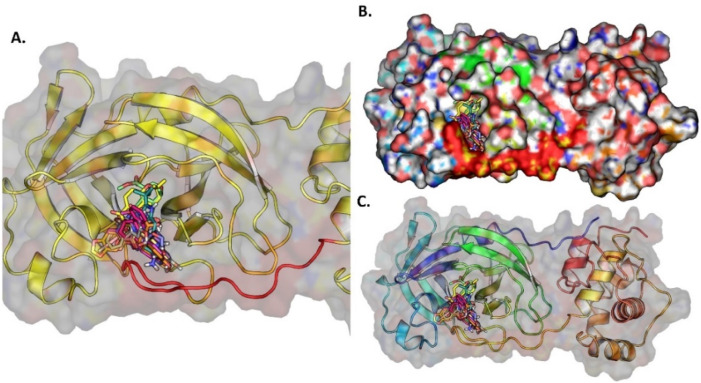
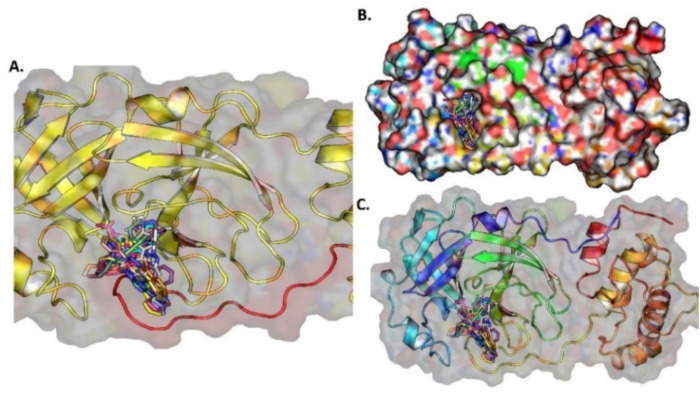
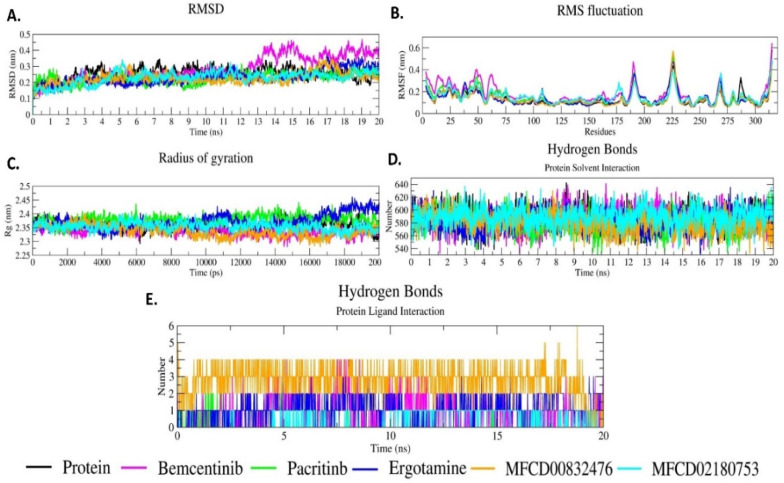
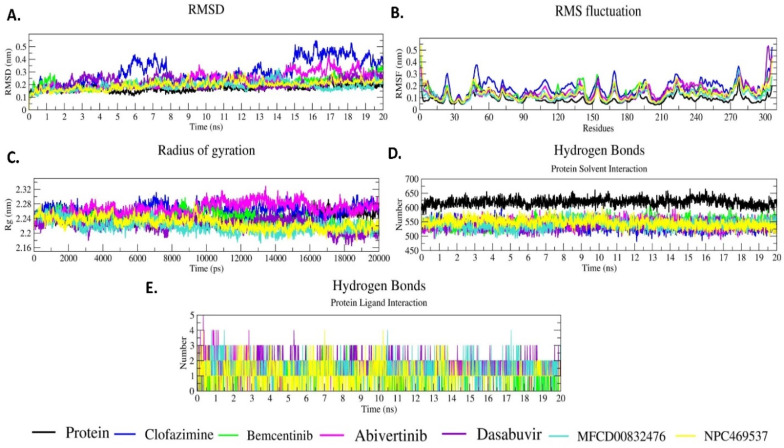
The pandemic, COVID-19, has spread worldwide and affected millions of people. There is an urgent need, therefore, to find a proper treatment for the novel coronavirus, Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), the causative agent. This paper focuses on identifying inhibitors that target SARS-CoV-2 proteases, PLPRO and 3CLPRO, which control the duplication and manages the life cycle of SARS-CoV-2. We have carried out detailed in silico Virtual high-throughput screening using Food and Drug Administration (FDA) approved drugs from the Zinc database, COVID-19 clinical trial compounds from Pubchem database, Natural compounds from Natural Product Activity and Species Source (NPASS) database and Maybridge database against PLPRO and 3CLPRO proteases. After thoroughly analyzing the screening results, we found five compounds, Bemcentinib, Pacritinib, Ergotamine, MFCD00832476, and MFCD02180753 inhibit PLPRO and six compounds, Bemcentinib, Clofazimine, Abivertinib, Dasabuvir, MFCD00832476, Leuconicine F inhibit the 3CLPRO. These compounds are stable within the protease proteins' active sites at 20ns MD simulation. The stability is revealed by hydrogen bond formations, hydrophobic interactions, and salt bridge interactions. Our study results also reveal that the selected five compounds against PLPRO and the six compounds against 3CLPRO bind to their active sites with good binding free energy. These compounds that inhibit the activity of PLPRO and 3CLPRO may, therefore, be used for treating COVID-19 infection.
Keywords: 3CL(PRO); COVID-19; Drug repositioning; PL(PRO); SARS-CoV-2.
Copyright © 2021 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
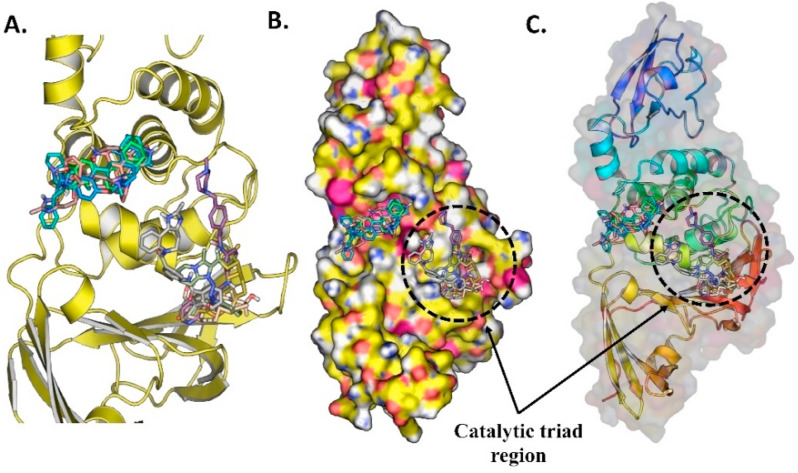
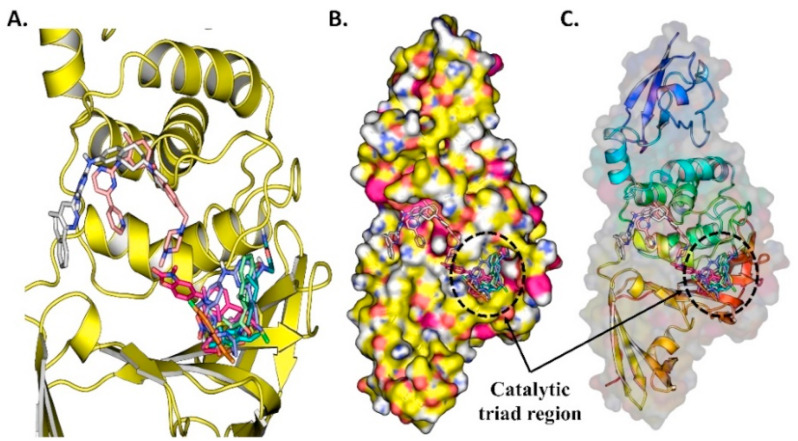
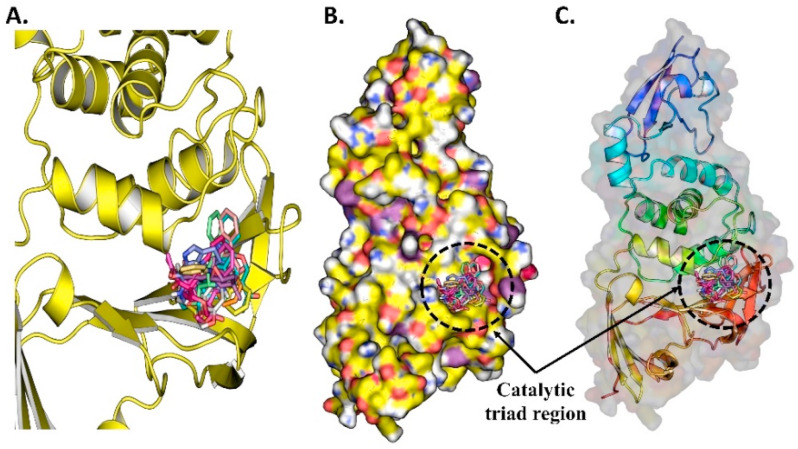
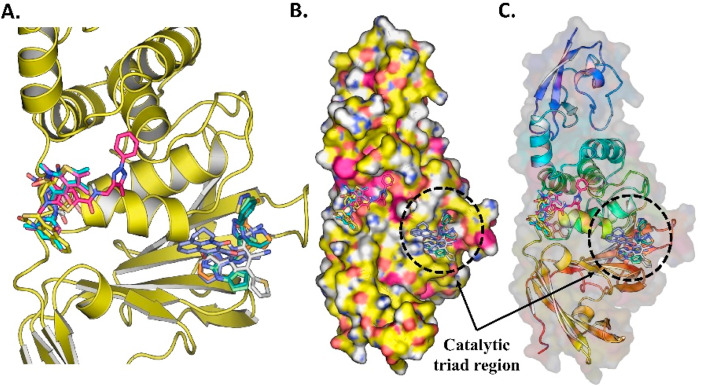
Similar articles
-
Repurposing FDA-approved drugs for COVID-19: targeting the main protease through multi-phase in silico approach.Antivir Ther. 2024 Dec;29(6):13596535241305536. doi: 10.1177/13596535241305536. Antivir Ther. 2024. PMID: 39639531
-
Screening potential FDA-approved inhibitors of the SARS-CoV-2 major protease 3CLpro through high-throughput virtual screening and molecular dynamics simulation.Aging (Albany NY). 2021 Mar 7;13(5):6258-6272. doi: 10.18632/aging.202703. Epub 2021 Mar 7. Aging (Albany NY). 2021. PMID: 33678621 Free PMC article.
-
Development of a Fluorescence-Based, High-Throughput SARS-CoV-2 3CLpro Reporter Assay.J Virol. 2020 Oct 27;94(22):e01265-20. doi: 10.1128/JVI.01265-20. Print 2020 Oct 27. J Virol. 2020. PMID: 32843534 Free PMC article.
-
Design and Evaluation of Anti-SARS-Coronavirus Agents Based on Molecular Interactions with the Viral Protease.Molecules. 2020 Aug 27;25(17):3920. doi: 10.3390/molecules25173920. Molecules. 2020. PMID: 32867349 Free PMC article. Review.
-
Identifying Exifone as a Dual-Target Agent Targeting Both SARS-CoV-2 3CL Protease and the ACE2/S-RBD Interaction Among Clinical Polyphenolic Compounds.Int J Mol Sci. 2025 Mar 2;26(5):2243. doi: 10.3390/ijms26052243. Int J Mol Sci. 2025. PMID: 40076865 Free PMC article. Review.
Cited by
-
Methodology-Centered Review of Molecular Modeling, Simulation, and Prediction of SARS-CoV-2.Chem Rev. 2022 Jul 13;122(13):11287-11368. doi: 10.1021/acs.chemrev.1c00965. Epub 2022 May 20. Chem Rev. 2022. PMID: 35594413 Free PMC article. Review.
-
The naturally-derived alkaloids as a potential treatment for COVID-19: A scoping review.Phytother Res. 2022 Jul;36(7):2686-2709. doi: 10.1002/ptr.7442. Epub 2022 Mar 30. Phytother Res. 2022. PMID: 35355337 Free PMC article.
-
Molecular docking as a tool for the discovery of novel insight about the role of acid sphingomyelinase inhibitors in SARS- CoV-2 infectivity.BMC Public Health. 2024 Feb 6;24(1):395. doi: 10.1186/s12889-024-17747-z. BMC Public Health. 2024. PMID: 38321448 Free PMC article. Review.
-
Potential Inhibitors Targeting Papain-Like Protease of SARS-CoV-2: Two Birds With One Stone.Front Chem. 2022 Feb 23;10:822785. doi: 10.3389/fchem.2022.822785. eCollection 2022. Front Chem. 2022. PMID: 35281561 Free PMC article. Review.
-
Hypericin Inhibit Alpha-Coronavirus Replication by Targeting 3CL Protease.Viruses. 2021 Sep 14;13(9):1825. doi: 10.3390/v13091825. Viruses. 2021. PMID: 34578406 Free PMC article.
References
-
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software. 2015;1:19–25.
-
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3clpro) structure: basis for design of anti-sars drugs. Science. 2003;300:1763. - PubMed
-
- Arya R., Das A., Prashar V., Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (Covid-19) from Fda approved drugs. Chemrxiv. 2020
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
