

PCIP-seq: simultaneous sequencing of integrated viral genomes and their insertion sites with long reads
- PMID: 33823910
- PMCID: PMC8025556
- DOI: 10.1186/s13059-021-02307-0
PCIP-seq: simultaneous sequencing of integrated viral genomes and their insertion sites with long reads
Abstract
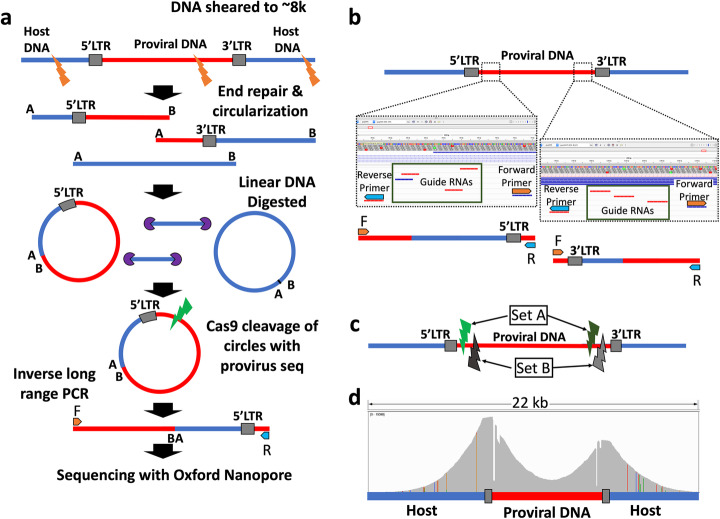
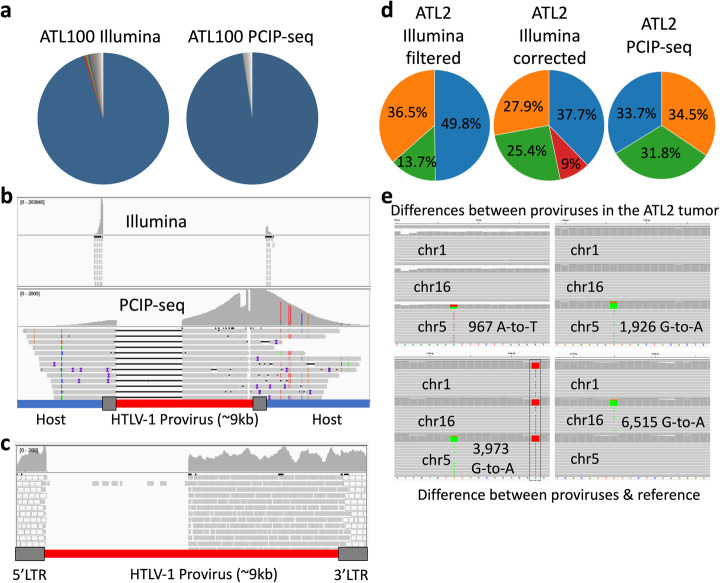
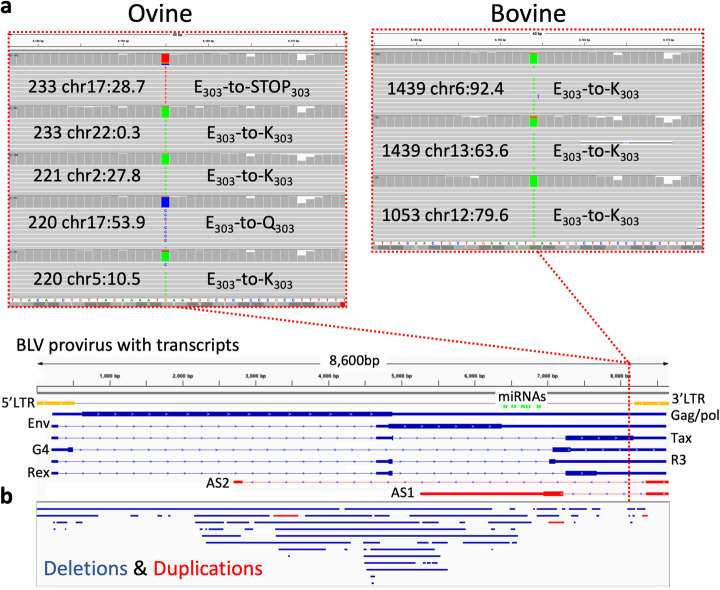
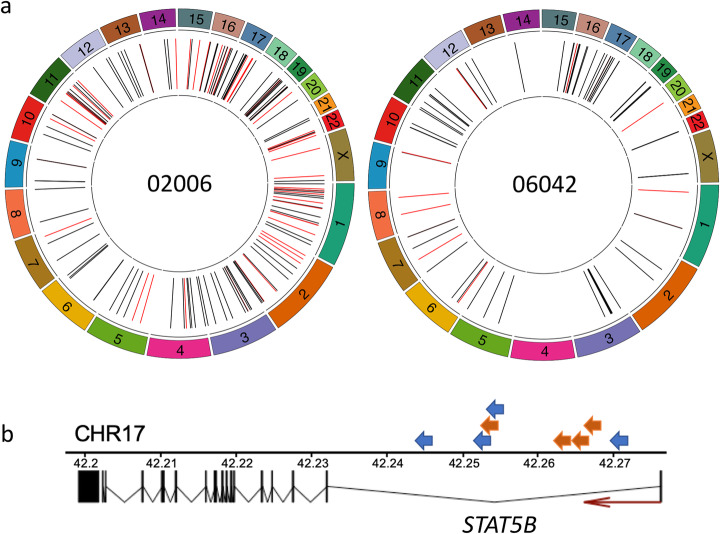
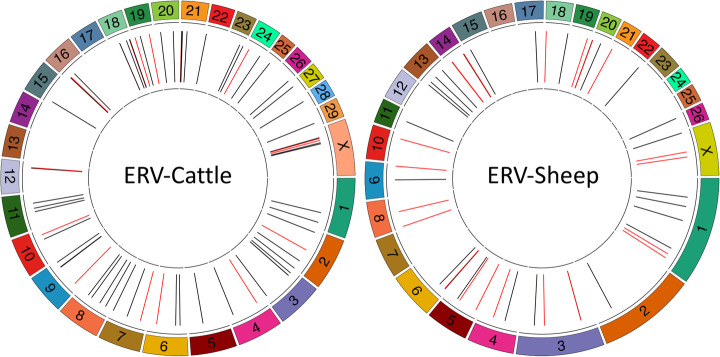
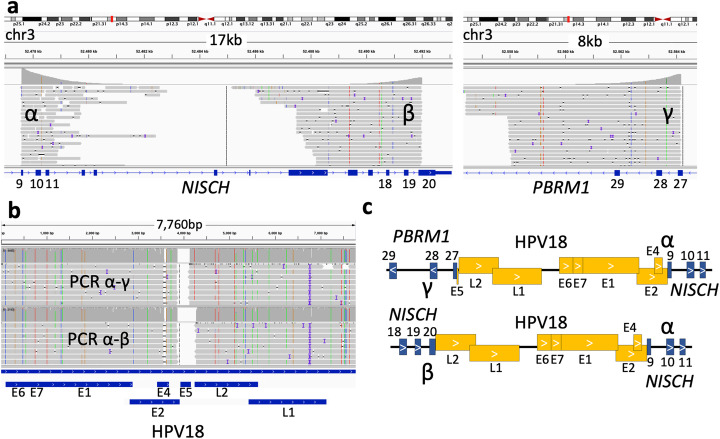
The integration of a viral genome into the host genome has a major impact on the trajectory of the infected cell. Integration location and variation within the associated viral genome can influence both clonal expansion and persistence of infected cells. Methods based on short-read sequencing can identify viral insertion sites, but the sequence of the viral genomes within remains unobserved. We develop PCIP-seq, a method that leverages long reads to identify insertion sites and sequence their associated viral genome. We apply the technique to exogenous retroviruses HTLV-1, BLV, and HIV-1, endogenous retroviruses, and human papillomavirus.
Keywords: BLV; Clonal expansion; HIV; HPV; HTLV-1; Integration site analysis; Long-read sequencing; NGS; Retrovirus; Viral genome.
Conflict of interest statement
The authors declare that they have no competing interests
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
