

Fibrocystic liver disease: novel concepts and translational perspectives
- PMID: 33824930
- PMCID: PMC7838530
- DOI: 10.21037/tgh-2020-04
Fibrocystic liver disease: novel concepts and translational perspectives
Abstract
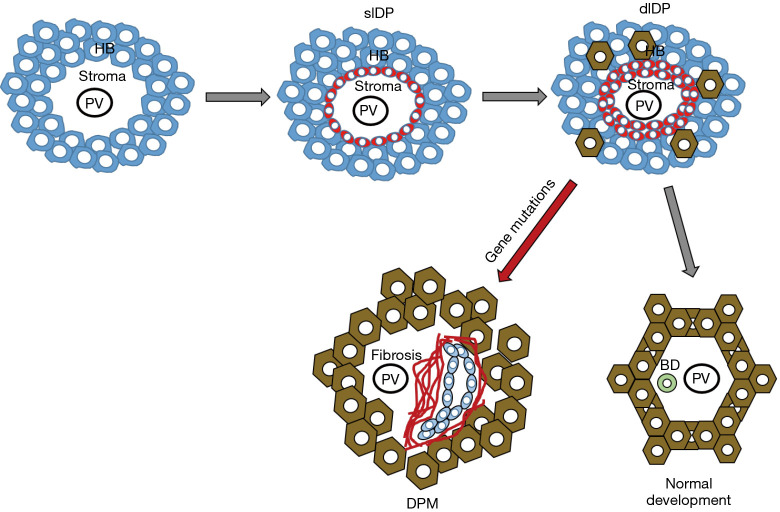
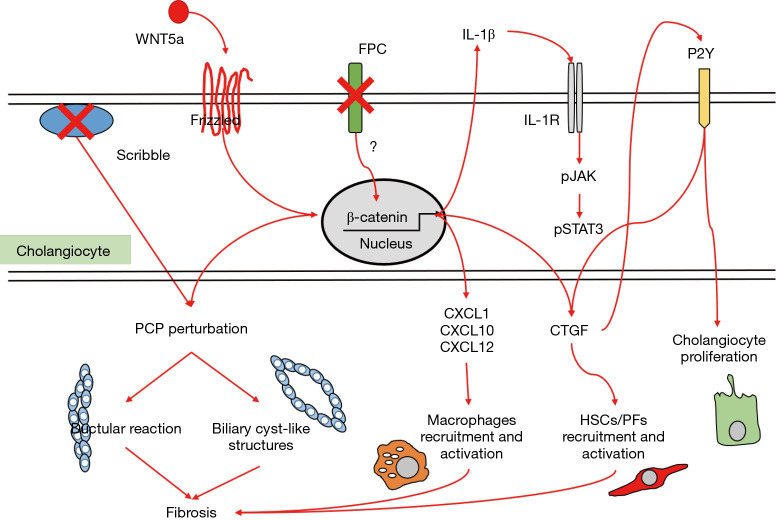
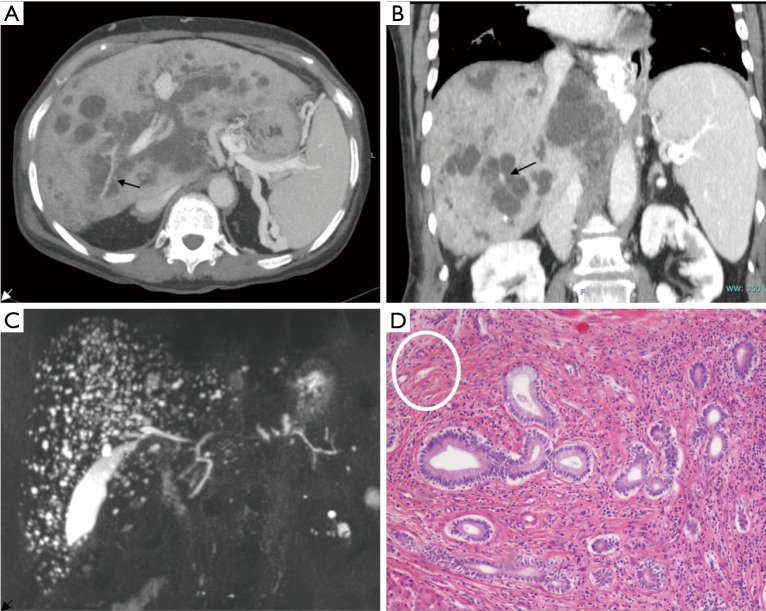
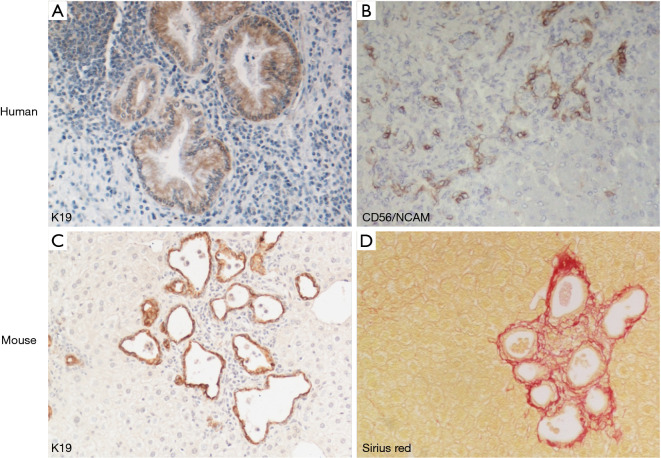
Fibrocystic liver diseases (FLDs) comprise a heterogeneous group of rare diseases of the biliary tree, having in common an abnormal development of the embryonic ductal plate caused by genetically-determined dysfunctions of proteins expressed in the primary cilia of cholangiocytes (and therefore grouped among the "ciliopathies"). The ductal dysgenesis may affect the biliary system at multiple levels, from the small intrahepatic bile ducts [congenital hepatic fibrosis (CHF)], to the larger intrahepatic bile ducts [Caroli disease (CD), or Caroli syndrome (CS), when CD coexists with CHF], leading to biliary microhamartomas and segmental bile duct dilations. Biliary changes are accompanied by progressive deposition of abundant peribiliary fibrosis. Peribiliary fibrosis and biliary cysts are the fundamental lesions of FLDs and are responsible for the main clinical manifestations, such as portal hypertension, recurrent cholangitis, cholestasis, sepsis and eventually cholangiocarcinoma. Furthermore, FLDs often associate with a spectrum of disorders affecting primarily the kidney. Among them, the autosomal recessive polycystic kidney disease (ARPKD) is the most frequent, and the renal function impairment is central in disease progression. CHF, CD/CS, and ARPKD are caused by a number of mutations in polycystic kidney hepatic disease 1 (PKHD1), a gene that encodes for fibrocystin/polyductin, a protein of unclear function, but supposedly involved in planar cell polarity and other fundamental cell functions. Targeted medical therapy is not available yet and thus the current treatment aims at controlling the complications. Interventional radiology or surgical treatments, including liver transplantation, are used in selected cases.
Keywords: Caroli disease (CD); Caroli syndrome (CS); Fibrocystic liver disease (FLD); biliary fibrosis; congenital hepatic fibrosis (CHF); polycystic kidney hepatic disease 1 (PKHD1).
2021 Translational Gastroenterology and Hepatology. All rights reserved.
Conflict of interest statement
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tgh-2020-04). The series “Recent Advances in Rare Liver Diseases” was commissioned by the editorial office without any funding or sponsorship. LF and MS served as the unpaid Guest Editors of the series. LF serves as an unpaid editorial board member of Translational Gastroenterology and Hepatology from Sep 2018 to Aug 2020. MS is a member of the advisory board of Esiai/Merk, Bayer, and Engitix, during the conduct of the study. The authors have no other conflicts of interest to declare.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources