

Thermodynamics and Free Energy Landscape of BAR-Domain Dimerization from Molecular Simulations
- PMID: 33826319
- PMCID: PMC8224822
- DOI: 10.1021/acs.jpcb.0c10992
Thermodynamics and Free Energy Landscape of BAR-Domain Dimerization from Molecular Simulations
Abstract
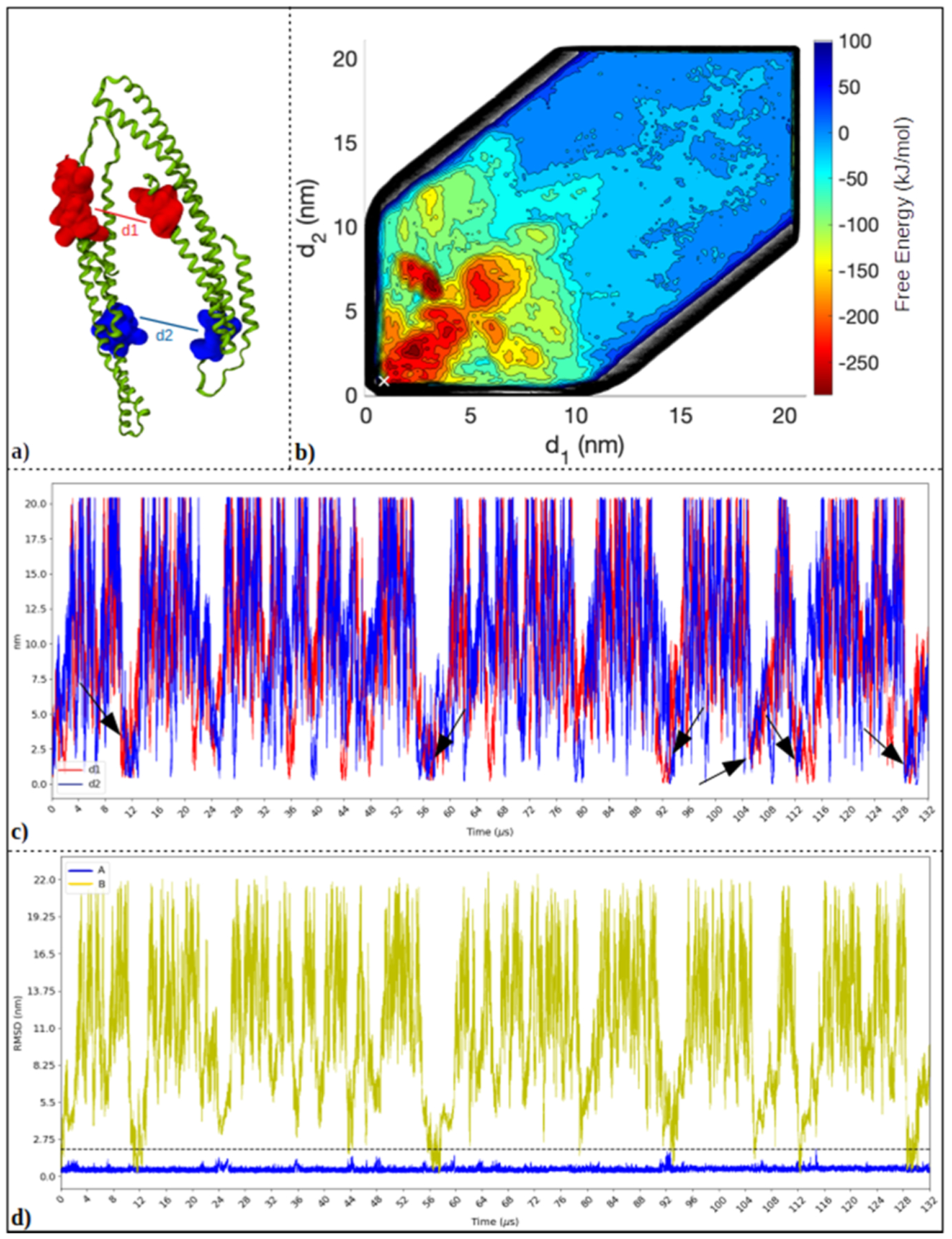
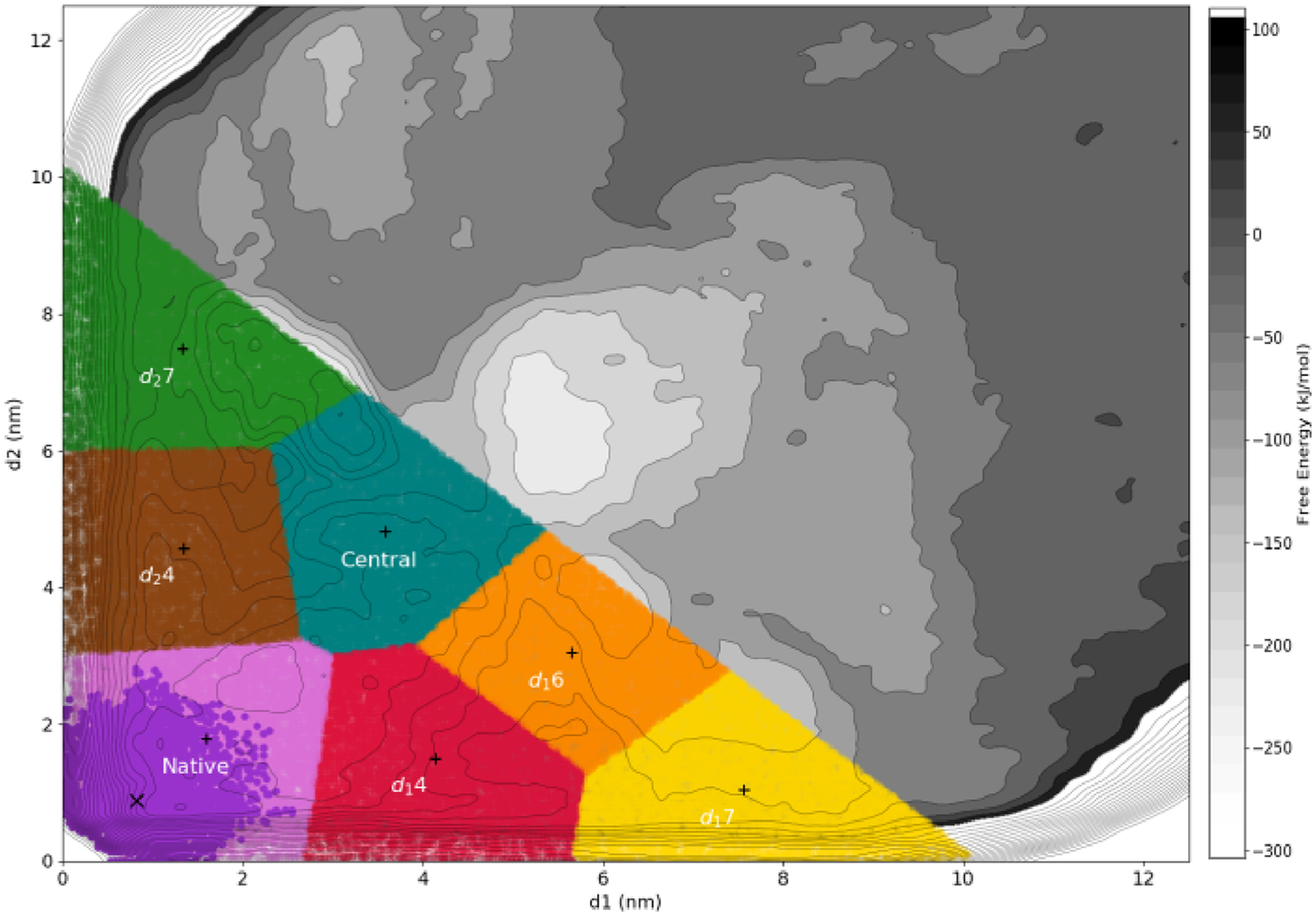
Proteins with BAR domains function to bind to and remodel biological membranes, where the dimerization of BAR domains is a key step in this function. These domains can dimerize in solution or after localizing to the membrane surface. Here, we characterize the binding thermodynamics of homodimerization between the LSP1 BAR domain proteins in solution, using molecular dynamics (MD) simulations. By combining the MARTINI coarse-grained protein models with enhanced sampling through metadynamics, we construct a two-dimensional free energy surface quantifying the bound versus unbound ensembles as a function of two distance variables. With this methodology, our simulations can simultaneously characterize the structures and relative stabilities of a range of sampled dimers, portraying a heterogeneous and extraordinarily stable bound ensemble, where the proper crystal structure dimer is the most stable in a 100 mM NaCl solution. Nonspecific dimers that are sampled involve contacts that are consistent with experimental structures of higher-order oligomers formed by the LSP1 BAR domain. Because the BAR dimers and oligomers can assemble on membranes, we characterize the relative alignment of the known membrane binding patches, finding that only the specific dimer is aligned to form strong interactions with the membrane. Hence, we would predict a strong selection of the specific dimer in binding to or assembling when on the membrane. Establishing the pairwise stabilities of homodimer contacts is difficult experimentally when the proteins form stable oligomers, but through the method used here, we can isolate these contacts, providing a foundation to study the same interactions on the membrane.
Conflict of interest statement
The authors declare no competing financial interest.
Figures






References
-
- Simunovic M; Evergren E; Callan-Jones A; Bassereau P Curving Cells Inside and Out: Roles of BAR Domain Proteins in Membrane Shaping and Its Cellular Implications. Annu. Rev. Cell Dev. Biol 2019, 35, 111–129. - PubMed
-
- McMahon HT; Boucrot E Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol 2011, 12, 517–533. - PubMed
-
- Ziółkowska NE; Karotki L; Rehman M; Huiskonen JT; Walther TC Eisosome-driven plasma membrane organization is mediated by BAR domains. Nat. Struct. Mol. Biol 2011, 18, 854–856. - PubMed
-
- Nicot AS; et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet 2007, 39, 1134–1139. - PubMed
-
- Masuda M; Mochizuki N Structural characteristics of BAR domain superfamily to sculpt the membrane. Semin. Cell Dev. Biol 2010, 21, 391–398. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
