

Identification of doxorubicin as a potential therapeutic against SARS-CoV-2 (COVID-19) protease: a molecular docking and dynamics simulation studies
- PMID: 33826483
- PMCID: PMC8043163
- DOI: 10.1080/07391102.2021.1905551
Identification of doxorubicin as a potential therapeutic against SARS-CoV-2 (COVID-19) protease: a molecular docking and dynamics simulation studies
Abstract
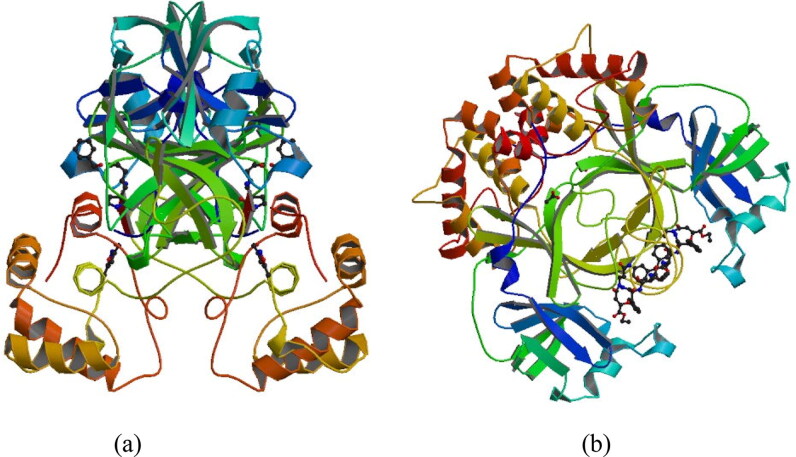
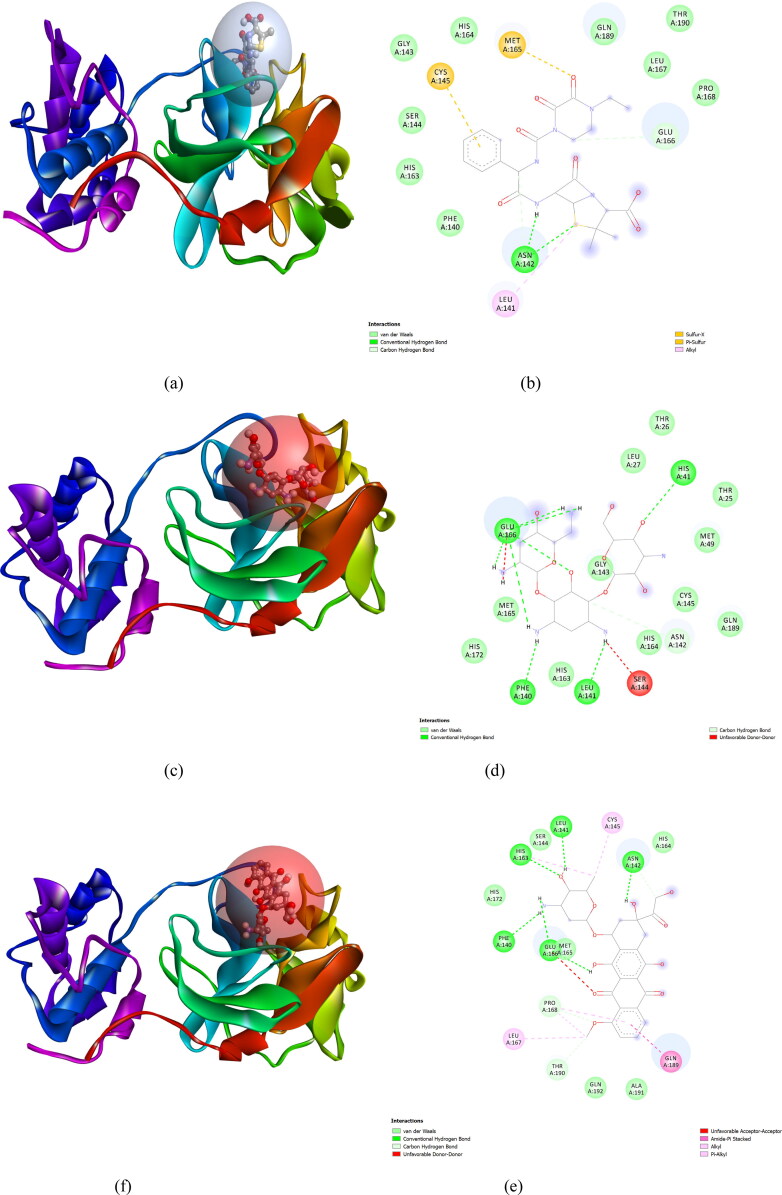
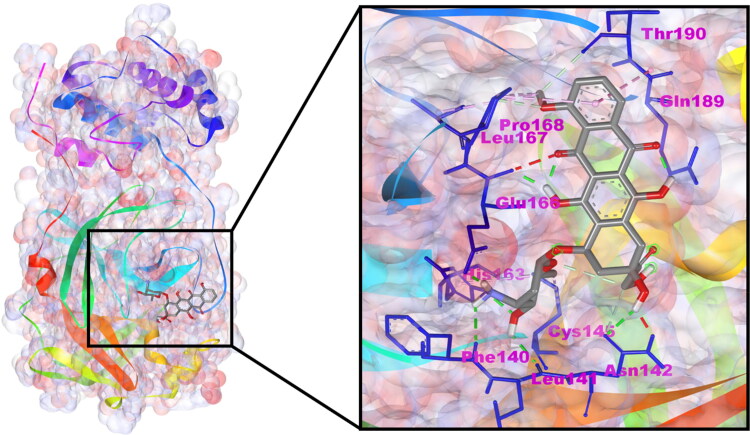
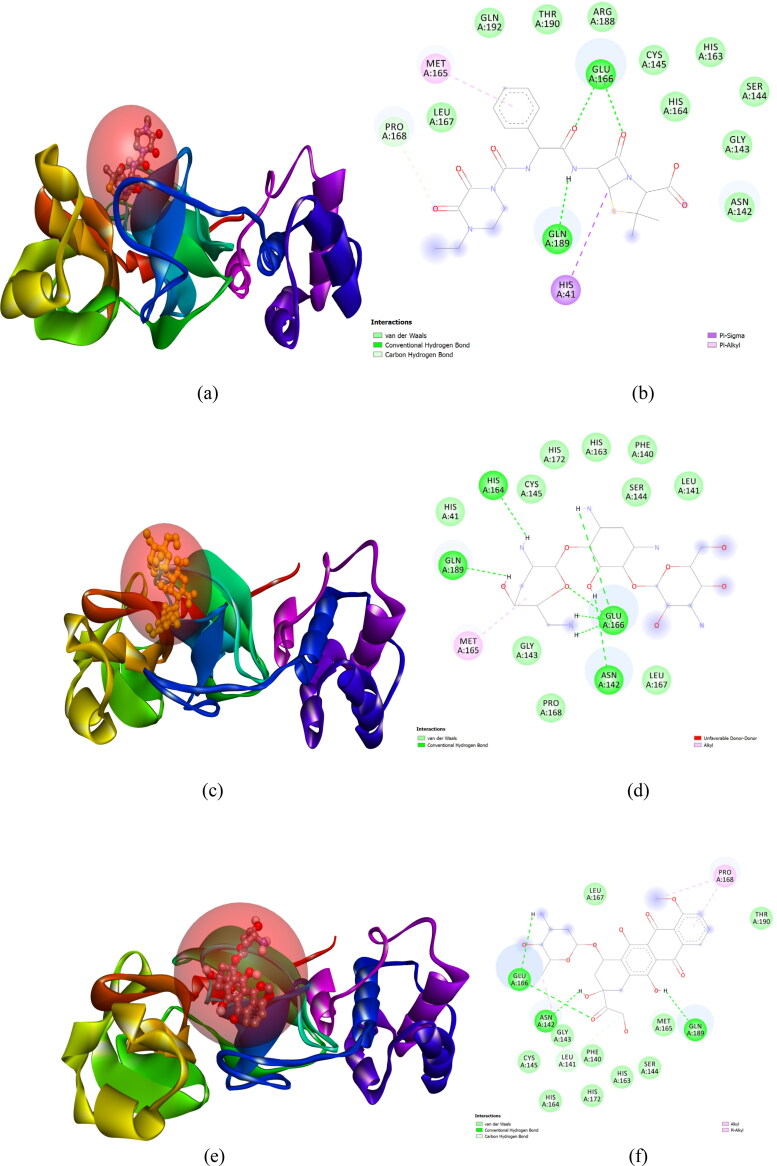
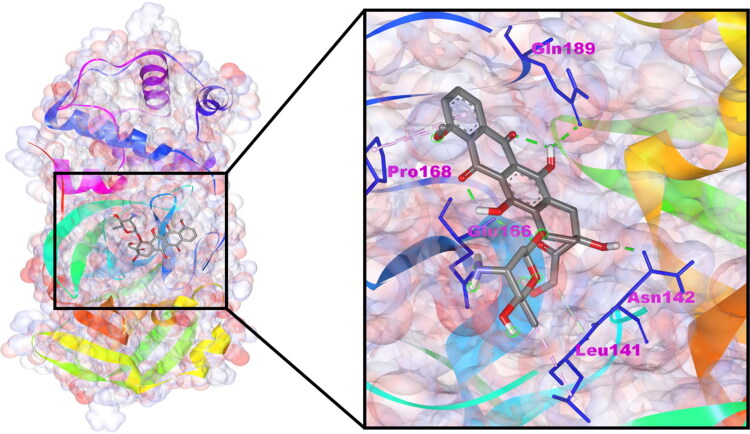
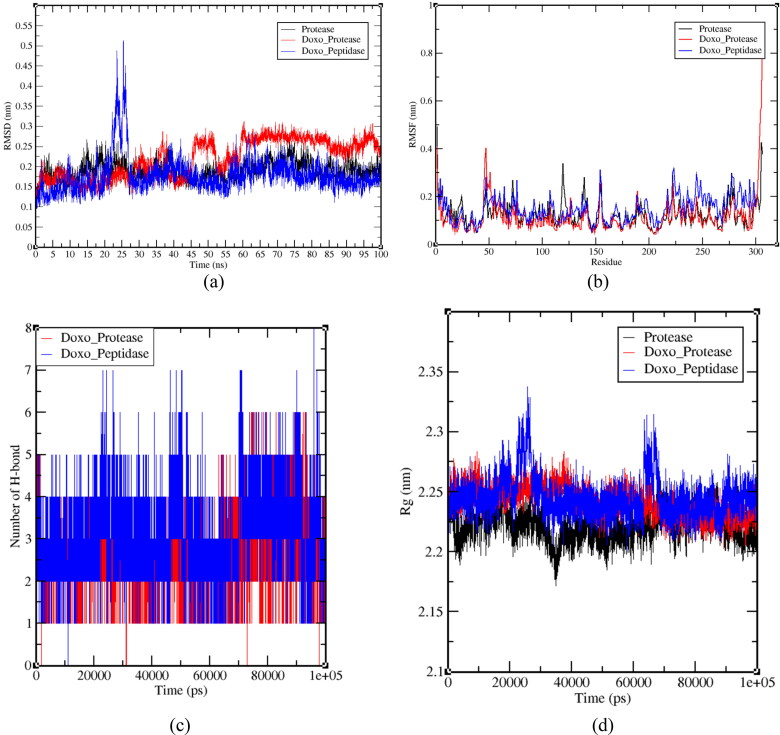
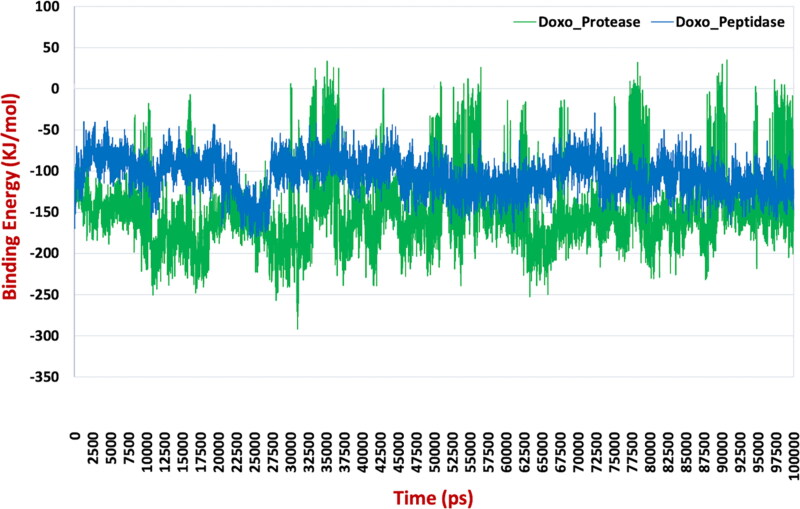
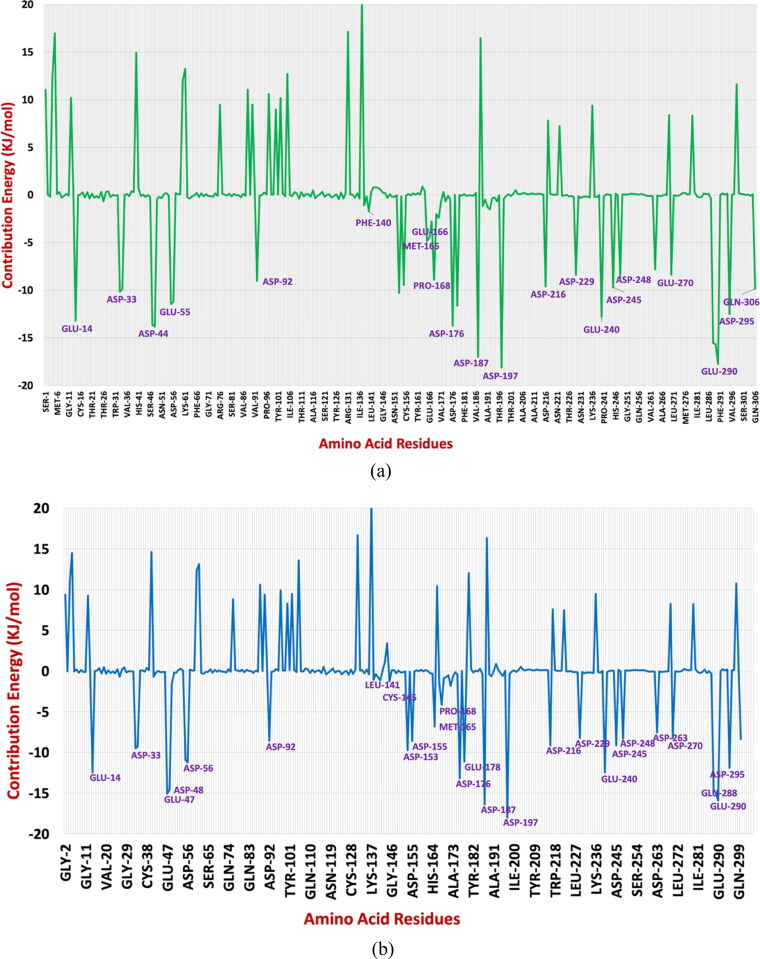
After one year, the COVID-19 pandemic caused by SARS-CoV-2 is still the largest concern for the scientific community. Of the many recognized drug targets of SARS-CoV-2, the main protease is one of the most important target due to its function in viral replication. We conducted an in silico study with repurposing drugs of antibiotics class against virus protease and peptidase using AutoDock tool. The following significant binding energy interaction was observed with protease (PDB: 6LU7) like piperacillin -7.25; tobramycin -9.20 and doxorubicin (Doxo) -10.04 kcal/mol and with peptidase (PDB: 2GTB) piperacillin -7.08; tobramycin -8.54 and Doxo -9.89 kcal/mol. Furthermore, the interaction and stability behavior of the Doxo-protease and Doxo-peptidase complexes were analyzed for a 100-nanosecond (ns) time. Calculated RMSD values observed using molecular dynamics simulation (MDS) were found to be 0.15-0.25 nm, RMSF calculation per residues showed a value near 0.2 nm and Rg values remained approximately 2.25 nm. MM-PBSA analysis of total binding energy calculation of Doxo-protease and Doxo-peptidase complexes are found to be -148.692 and -105.367 kJ/mol, respectively. Moreover, amino acid residue ASP-197 showed the lowest contribution binding energy i.e. -18.1185 kJ/mol, and amino acid residue ASP-187 showed -17.0267 kJ/mol contribution energy. Thus, significant docking interaction and stable dynamicity of Doxo-protease complex with time was suggested that Doxo could be a choice to inhibit potentially the viral proteases that could prevent the entry inside the host cell to control the COVID-19 disease. Communicated by Ramaswamy H. Sarma.
Keywords: Doxorubicin; MDS; SARS-CoV-2; antiviral drugs; protease inhibitor.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Figures
Similar articles
-
Identification of natural inhibitors against Mpro of SARS-CoV-2 by molecular docking, molecular dynamics simulation, and MM/PBSA methods.J Biomol Struct Dyn. 2022 Apr;40(6):2757-2768. doi: 10.1080/07391102.2020.1842806. Epub 2020 Nov 4. J Biomol Struct Dyn. 2022. PMID: 33143552 Free PMC article.
-
Molecular Binding Mechanism and Pharmacology Comparative Analysis of Noscapine for Repurposing against SARS-CoV-2 Protease.J Proteome Res. 2020 Nov 6;19(11):4678-4689. doi: 10.1021/acs.jproteome.0c00367. Epub 2020 Sep 4. J Proteome Res. 2020. PMID: 32786685
-
Binding and inhibitory effect of ravidasvir on 3CLpro of SARS-CoV-2: a molecular docking, molecular dynamics and MM/PBSA approach.J Biomol Struct Dyn. 2022 Oct;40(16):7303-7310. doi: 10.1080/07391102.2021.1896388. Epub 2021 Mar 8. J Biomol Struct Dyn. 2022. PMID: 33682639
-
Possibility of HIV-1 protease inhibitors-clinical trial drugs as repurposed drugs for SARS-CoV-2 main protease: a molecular docking, molecular dynamics and binding free energy simulation study.J Biomol Struct Dyn. 2021 Sep;39(15):5368-5375. doi: 10.1080/07391102.2020.1786459. Epub 2020 Jul 6. J Biomol Struct Dyn. 2021. PMID: 32627689 Free PMC article.
-
Methylxanthines as Potential Inhibitor of SARS-CoV-2: an In Silico Approach.Curr Pharmacol Rep. 2022;8(2):149-170. doi: 10.1007/s40495-021-00276-3. Epub 2022 Mar 8. Curr Pharmacol Rep. 2022. PMID: 35281252 Free PMC article. Review.
Cited by
-
Exploring the Binding Interaction of Active Compound of Pineapple against Foodborne Bacteria and Novel Coronavirus (SARS-CoV-2) Based on Molecular Docking and Simulation Studies.Nutrients. 2022 Jul 25;14(15):3045. doi: 10.3390/nu14153045. Nutrients. 2022. PMID: 35893899 Free PMC article.
-
Pea eggplant (Solanum torvum Swartz) is a source of plant food polyphenols with SARS-CoV inhibiting potential.PeerJ. 2022 Nov 29;10:e14168. doi: 10.7717/peerj.14168. eCollection 2022. PeerJ. 2022. PMID: 36518265 Free PMC article.
-
Discovery of new drug indications for COVID-19: A drug repurposing approach.PLoS One. 2022 May 24;17(5):e0267095. doi: 10.1371/journal.pone.0267095. eCollection 2022. PLoS One. 2022. PMID: 35609015 Free PMC article.
-
The Nucleolus and Its Interactions with Viral Proteins Required for Successful Infection.Cells. 2024 Sep 21;13(18):1591. doi: 10.3390/cells13181591. Cells. 2024. PMID: 39329772 Free PMC article. Review.
-
In silico identification of potential inhibitors of vital monkeypox virus proteins from FDA approved drugs.Mol Divers. 2023 Oct;27(5):2169-2184. doi: 10.1007/s11030-022-10550-1. Epub 2022 Nov 4. Mol Divers. 2023. PMID: 36331784 Free PMC article.
References
-
- Ahmad, V. (2020)., Prospective of extracellular matrix and drug correlations in disease management. Asian Journal of Pharmaceutical Sciences, ISSN 1818–0876, 10.1016/j.ajps.2020.06.007. (https://www.sciencedirect.com/science/article/pii/S181808761931520X) - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous