

Optimized permutation testing for information theoretic measures of multi-gene interactions
- PMID: 33827420
- PMCID: PMC8028212
- DOI: 10.1186/s12859-021-04107-6
Optimized permutation testing for information theoretic measures of multi-gene interactions
Abstract
Background: Permutation testing is often considered the "gold standard" for multi-test significance analysis, as it is an exact test requiring few assumptions about the distribution being computed. However, it can be computationally very expensive, particularly in its naive form in which the full analysis pipeline is re-run after permuting the phenotype labels. This can become intractable in multi-locus genome-wide association studies (GWAS), in which the number of potential interactions to be tested is combinatorially large.
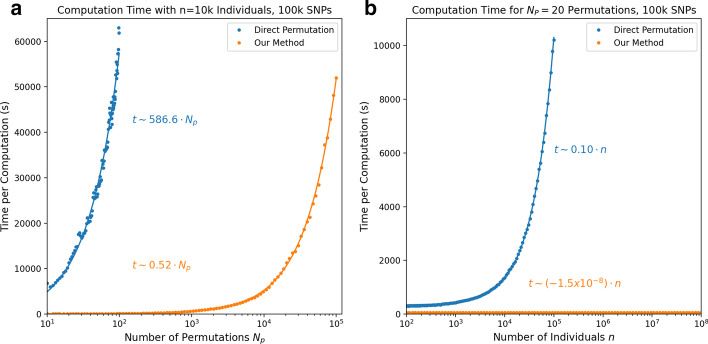
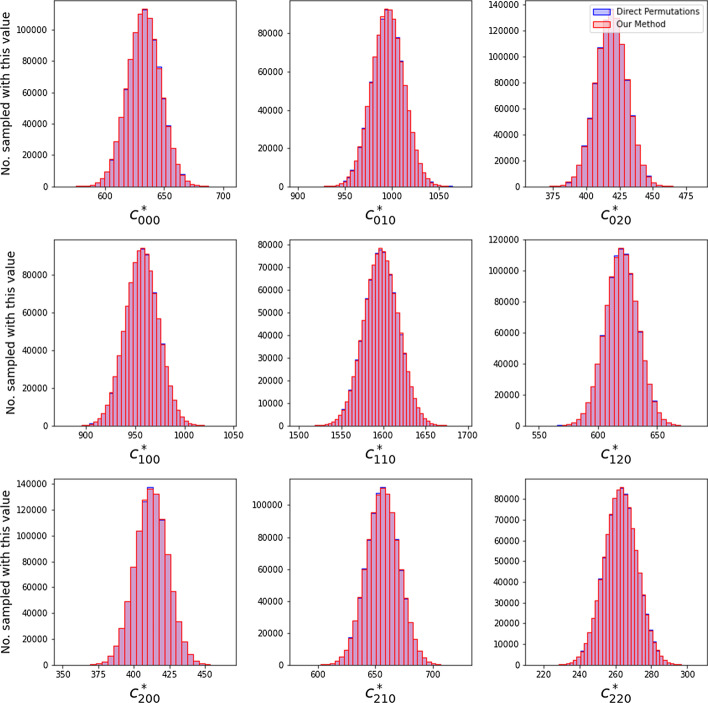
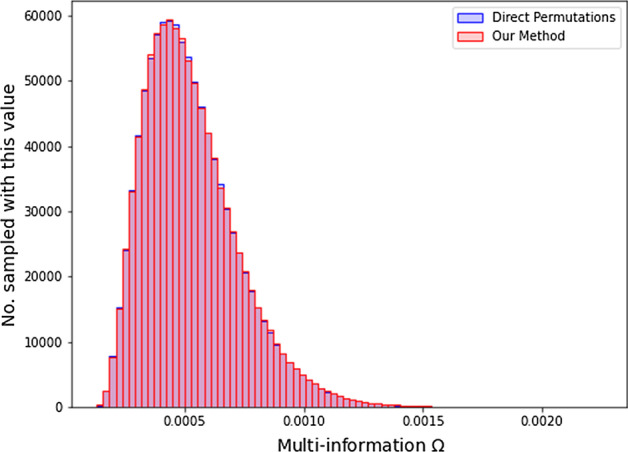
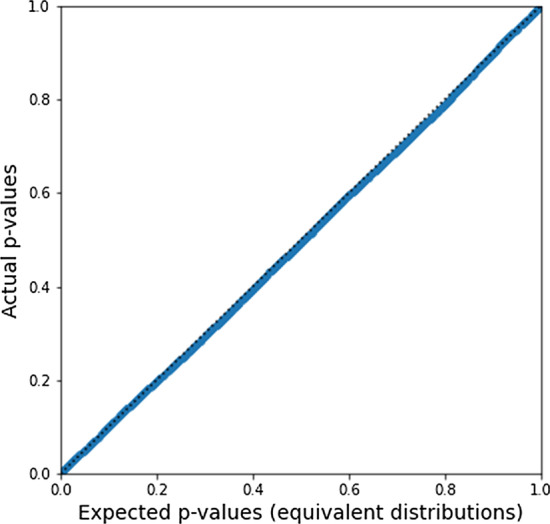
Results: In this paper, we develop an approach for permutation testing in multi-locus GWAS, specifically focusing on SNP-SNP-phenotype interactions using multivariable measures that can be computed from frequency count tables, such as those based in Information Theory. We find that the computational bottleneck in this process is the construction of the count tables themselves, and that this step can be eliminated at each iteration of the permutation testing by transforming the count tables directly. This leads to a speed-up by a factor of over 103 for a typical permutation test compared to the naive approach. Additionally, this approach is insensitive to the number of samples making it suitable for datasets with large number of samples.
Conclusions: The proliferation of large-scale datasets with genotype data for hundreds of thousands of individuals enables new and more powerful approaches for the detection of multi-locus genotype-phenotype interactions. Our approach significantly improves the computational tractability of permutation testing for these studies. Moreover, our approach is insensitive to the large number of samples in these modern datasets. The code for performing these computations and replicating the figures in this paper is freely available at https://github.com/kunert/permute-counts .
Keywords: Information theory; Multi-locus GWAS; Multivariable interactions; Permutation testing.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
