

SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation
- PMID: 33827897
- PMCID: PMC8139422
- DOI: 10.1126/sciimmunol.abg0833
SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation
Abstract
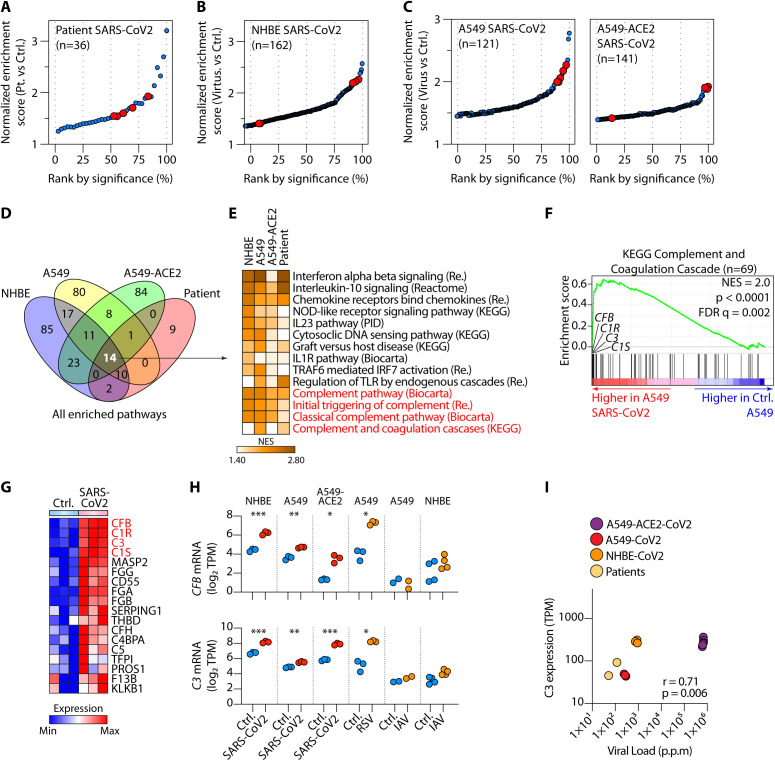
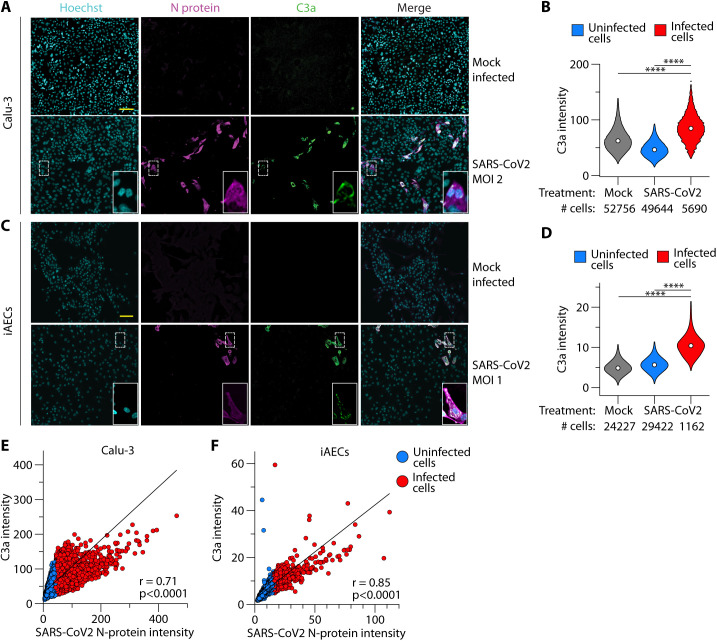
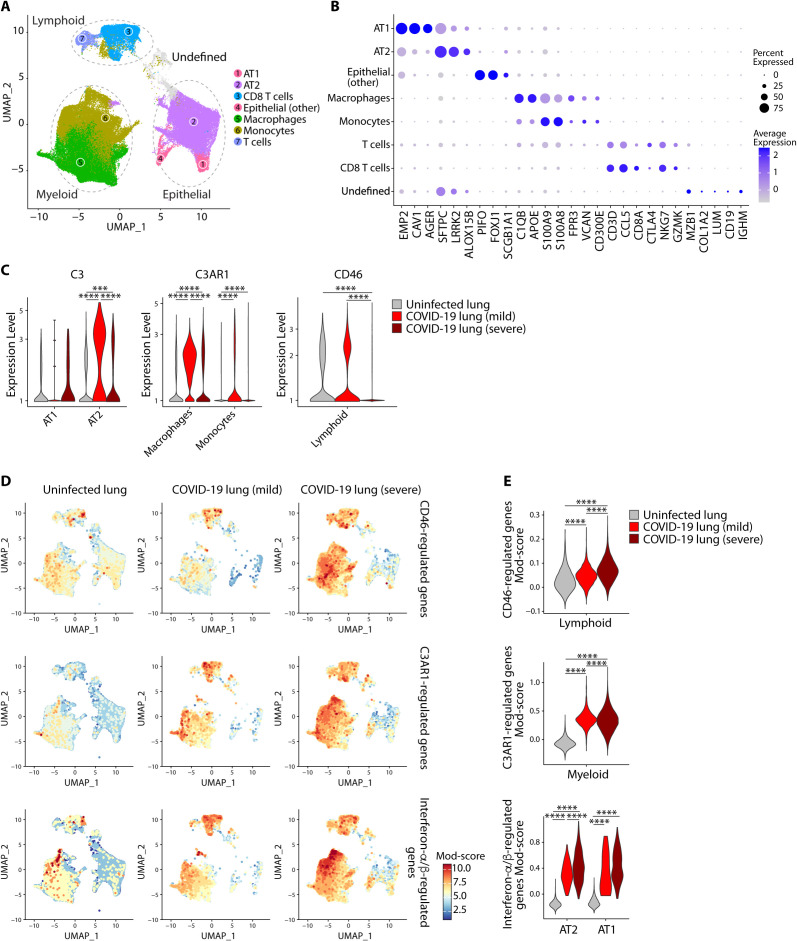
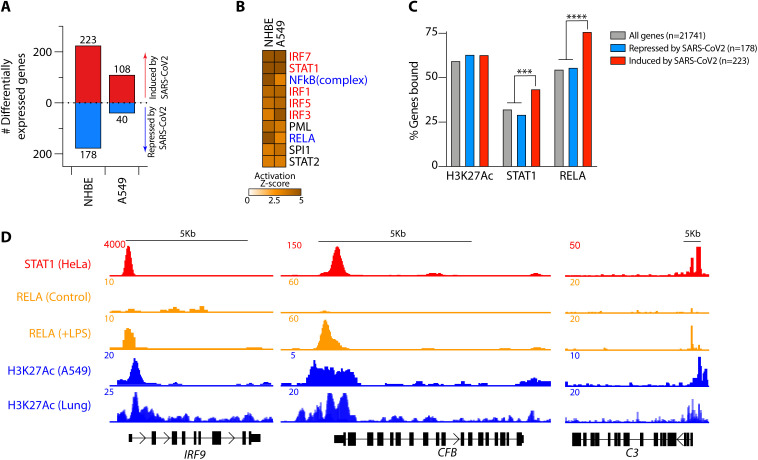
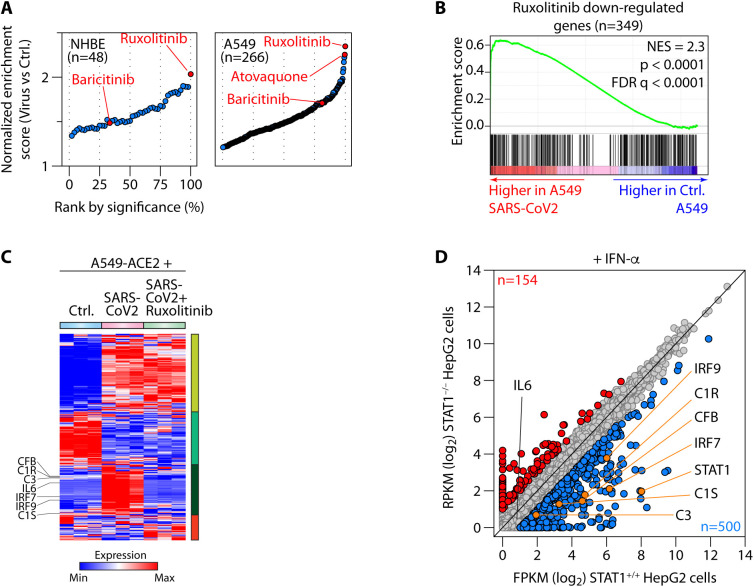
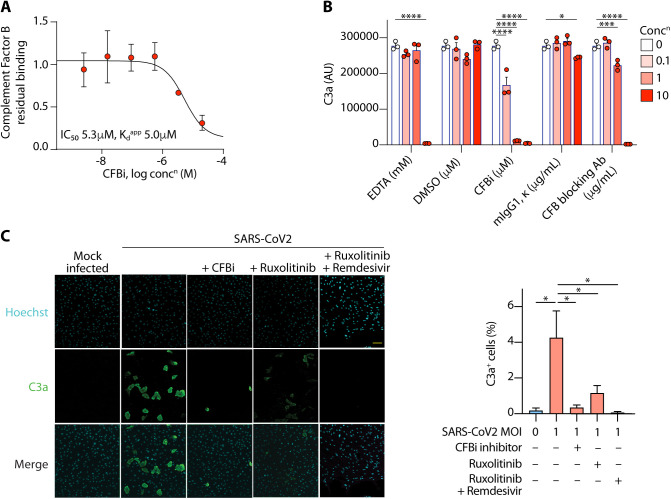
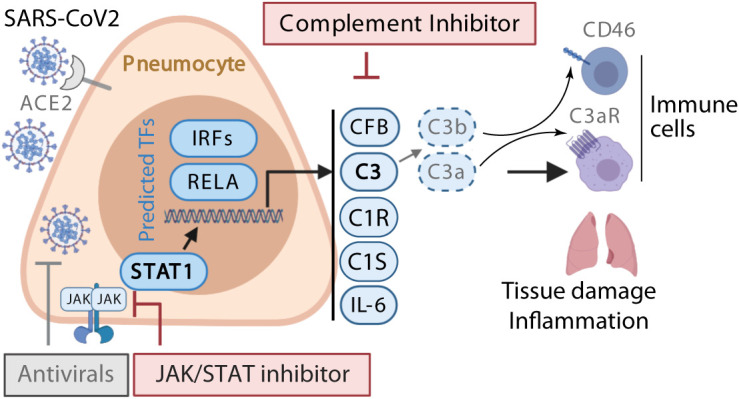
Patients with coronavirus disease 2019 (COVID-19) present a wide range of acute clinical manifestations affecting the lungs, liver, kidneys and gut. Angiotensin converting enzyme (ACE) 2, the best-characterized entry receptor for the disease-causing virus SARS-CoV-2, is highly expressed in the aforementioned tissues. However, the pathways that underlie the disease are still poorly understood. Here, we unexpectedly found that the complement system was one of the intracellular pathways most highly induced by SARS-CoV-2 infection in lung epithelial cells. Infection of respiratory epithelial cells with SARS-CoV-2 generated activated complement component C3a and could be blocked by a cell-permeable inhibitor of complement factor B (CFBi), indicating the presence of an inducible cell-intrinsic C3 convertase in respiratory epithelial cells. Within cells of the bronchoalveolar lavage of patients, distinct signatures of complement activation in myeloid, lymphoid and epithelial cells tracked with disease severity. Genes induced by SARS-CoV-2 and the drugs that could normalize these genes both implicated the interferon-JAK1/2-STAT1 signaling system and NF-κB as the main drivers of their expression. Ruxolitinib, a JAK1/2 inhibitor, normalized interferon signature genes and all complement gene transcripts induced by SARS-CoV-2 in lung epithelial cell lines, but did not affect NF-κB-regulated genes. Ruxolitinib, alone or in combination with the antiviral remdesivir, inhibited C3a protein produced by infected cells. Together, we postulate that combination therapy with JAK inhibitors and drugs that normalize NF-κB-signaling could potentially have clinical application for severe COVID-19.
Copyright © 2021, American Association for the Advancement of Science.
Figures
Update of
-
SARS-CoV2 drives JAK1/2-dependent local and systemic complement hyper-activation.Res Sq [Preprint]. 2020 Jun 9:rs.3.rs-33390. doi: 10.21203/rs.3.rs-33390/v1. Res Sq. 2020. Update in: Sci Immunol. 2021 Apr 7;6(58):eabg0833. doi: 10.1126/sciimmunol.abg0833. PMID: 32702726 Free PMC article. Updated. Preprint.
Comment in
-
Complement control for COVID-19.Sci Immunol. 2021 May 25;6(59):eabj1014. doi: 10.1126/sciimmunol.abj1014. Sci Immunol. 2021. PMID: 34035117 Free PMC article.
References
-
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X., Guan L., Wei Y., Li H., Wu X., Xu J., Tu S., Zhang Y., Chen H., Cao B., Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 395, 1054–1062. (2020). 10.1016/S0140-6736(20)30566-3 - DOI - PMC - PubMed
-
- Mandal S., Barnett J., Brill S. E., Brown J. S., Denneny E. K., Hare S. S., Heightman M., Hillman T. E., Jacob J., Jarvis H. C., Lipman M. C. I., Naidu S. B., Nair A., Porter J. C., Tomlinson G. S., Hurst J. R., ARC Study Group , Long-COVID: A cross-sectional study of persisting symptoms, biomarker and imaging abnormalities following hospitalisation for COVID-19. Thorax 76, 396–398. (2021). 10.1136/thoraxjnl-2020-215818 - DOI - PMC - PubMed
-
- Cardone J., Le Friec G., Vantourout P., Roberts A., Fuchs A., Jackson I., Suddason T., Lord G., Atkinson J. P., Cope A., Hayday A., Kemper C., Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat. Immunol. 11, 862–871. (2010). 10.1038/ni.1917 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
