

Signaling Pathways Involved in Nutrient Sensing Control in Cancer Stem Cells: An Overview
- PMID: 33828530
- PMCID: PMC8020906
- DOI: 10.3389/fendo.2021.627745
Signaling Pathways Involved in Nutrient Sensing Control in Cancer Stem Cells: An Overview
Abstract
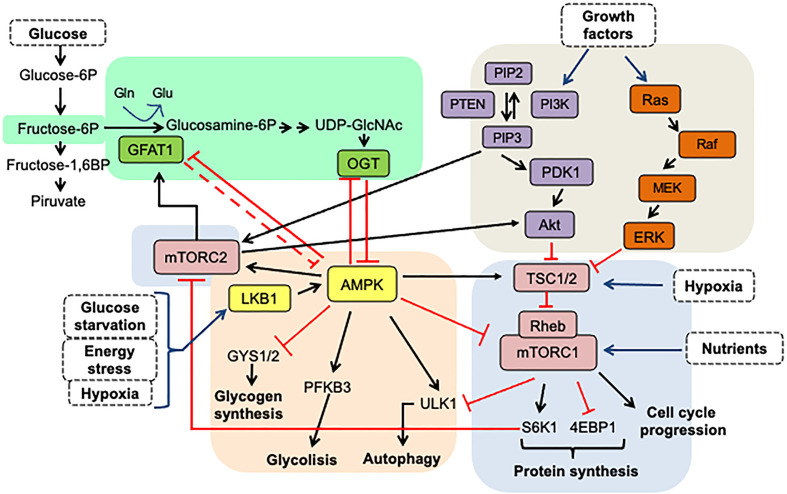
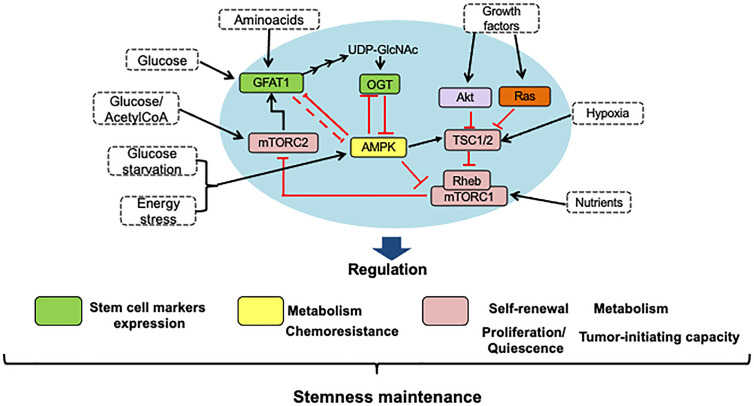
Cancer cells characteristically have a high proliferation rate. Because tumor growth depends on energy-consuming anabolic processes, including biosynthesis of protein, lipid, and nucleotides, many tumor-associated conditions, including intermittent oxygen deficiency due to insufficient vascularization, oxidative stress, and nutrient deprivation, results from fast growth. To cope with these environmental stressors, cancer cells, including cancer stem cells, must adapt their metabolism to maintain cellular homeostasis. It is well- known that cancer stem cells (CSC) reprogram their metabolism to adapt to live in hypoxic niches. They usually change from oxidative phosphorylation to increased aerobic glycolysis even in the presence of oxygen. However, as opposed to most differentiated cancer cells relying on glycolysis, CSCs can be highly glycolytic or oxidative phosphorylation-dependent, displaying high metabolic plasticity. Although the influence of the metabolic and nutrient-sensing pathways on the maintenance of stemness has been recognized, the molecular mechanisms that link these pathways to stemness are not well known. Here in this review, we describe the most relevant signaling pathways involved in nutrient sensing and cancer cell survival. Among them, Adenosine monophosphate (AMP)-activated protein kinase (AMPK) pathway, mTOR pathway, and Hexosamine Biosynthetic Pathway (HBP) are critical sensors of cellular energy and nutrient status in cancer cells and interact in complex and dynamic ways.
Keywords: adenosine monophosphate-activated protein kinase (AMPK) signaling; cancer stem cells; hexosamine biosynthesis pathway (HBP) pathway; mammalian target of rapamycin (mTOR) signaling; nutrient sensing.
Copyright © 2021 Robles-Flores, Moreno-Londoño and Castañeda-Patlán.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional perspective of diabetes, obesity, and cancer.Sci STKE. 2006 Aug 1;2006(346):re7. doi: 10.1126/stke.3462006re7. Sci STKE. 2006. PMID: 16885148 Review.
-
Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway.Cell Cycle. 2013 Sep 15;12(18):3109-24. doi: 10.4161/cc.26173. Epub 2013 Aug 21. Cell Cycle. 2013. PMID: 23974095 Free PMC article.
-
Cross-talk between AMPK and mTOR in regulating energy balance.Crit Rev Food Sci Nutr. 2012;52(5):373-81. doi: 10.1080/10408398.2010.500245. Crit Rev Food Sci Nutr. 2012. PMID: 22369257 Review.
-
Sensing of nutrients and microbes in the gut.Curr Opin Gastroenterol. 2016 Mar;32(2):86-95. doi: 10.1097/MOG.0000000000000246. Curr Opin Gastroenterol. 2016. PMID: 26836123 Review.
-
PAS kinase is a nutrient and energy sensor in hypothalamic areas required for the normal function of AMPK and mTOR/S6K1.Mol Neurobiol. 2014 Oct;50(2):314-26. doi: 10.1007/s12035-013-8630-4. Epub 2014 Jan 21. Mol Neurobiol. 2014. PMID: 24445950
Cited by
-
Dietary Restrictions and Cancer Prevention: State of the Art.Nutrients. 2025 Jan 29;17(3):503. doi: 10.3390/nu17030503. Nutrients. 2025. PMID: 39940361 Free PMC article. Review.
-
mTOR signalling pathway in stem cell bioactivities and angiogenesis potential.Cell Prolif. 2023 Dec;56(12):e13499. doi: 10.1111/cpr.13499. Epub 2023 May 8. Cell Prolif. 2023. PMID: 37156724 Free PMC article. Review.
-
Genome Scale Modeling to Study the Metabolic Competition between Cells in the Tumor Microenvironment.Cancers (Basel). 2021 Sep 14;13(18):4609. doi: 10.3390/cancers13184609. Cancers (Basel). 2021. PMID: 34572839 Free PMC article. Review.
-
The Role of Metabolism in the Development of Personalized Therapies in Acute Myeloid Leukemia.Front Oncol. 2021 May 19;11:665291. doi: 10.3389/fonc.2021.665291. eCollection 2021. Front Oncol. 2021. PMID: 34094959 Free PMC article. Review.
-
The metabolomic landscape plays a critical role in glioma oncogenesis.Cancer Sci. 2022 May;113(5):1555-1563. doi: 10.1111/cas.15325. Epub 2022 Mar 23. Cancer Sci. 2022. PMID: 35271755 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous