

Novel opportunities for NGS-based one health surveillance of foodborne viruses
- PMID: 33829135
- PMCID: PMC7993515
- DOI: 10.1186/s42522-020-00015-6
Novel opportunities for NGS-based one health surveillance of foodborne viruses
Abstract
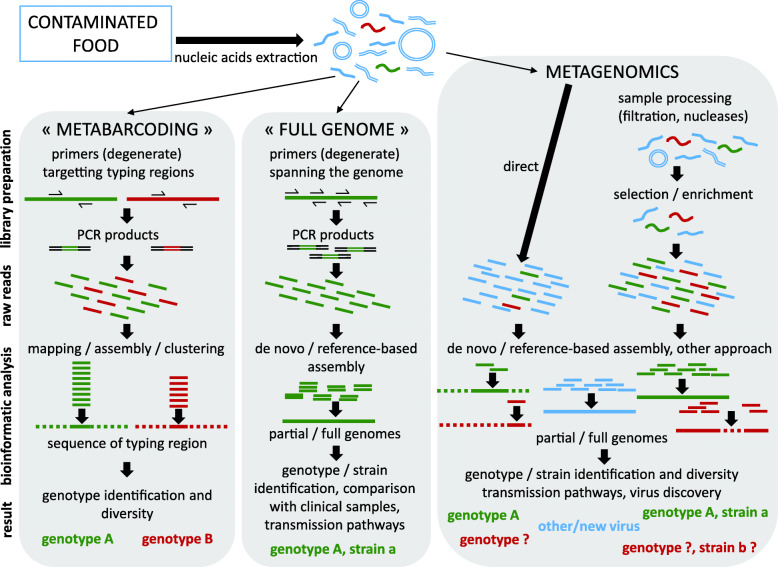
Foodborne viral infections rank among the top 5 causes of disease, with noroviruses and hepatitis A causing the greatest burden globally. Contamination of foods by infected food handlers or through environmental pollution are the main sources of foodborne illness, with a lesser role for consumption of products from infected animals. Viral partial genomic sequencing has been used for more than two decades to track foodborne outbreaks and whole genome or metagenomics next-generation-sequencing (NGS) are new additions to the toolbox of food microbiology laboratories. We discuss developments in the field of targeted and metagenomic NGS, with an emphasis on application in food virology, the challenges and possible solutions towards future routine application.
Keywords: Food virology; Foodborne virus; Human enteric virus; Metagenomics; Next-generation sequencing; Norovirus.
© The Author(s) 2020.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures
References
-
- Izopet, J, P Tremeaux, O Marion, M Migueres, N Capelli, S Chapuy-Regaud, et al.; Hepatitis e virus infections in europe. J Clin Virol. 2019;120(20–26. - PubMed
Publication types
LinkOut - more resources
Full Text Sources