

High-Resolution Structures of K+ Channels
- PMID: 33829342
- PMCID: PMC9382700
- DOI: 10.1007/164_2021_454
High-Resolution Structures of K+ Channels
Abstract
Potassium channels are present in every living cell and essential to setting up a stable, non-zero transmembrane electrostatic potential which manifests the off-equilibrium livelihood of the cell. They are involved in other cellular activities and regulation, such as the controlled release of hormones, the activation of T-cells for immune response, the firing of action potential in muscle cells and neurons, etc. Pharmacological reagents targeting potassium channels are important for treating various human diseases linked to dysfunction of the channels. High-resolution structures of these channels are very useful tools for delineating the detailed chemical basis underlying channel functions and for structure-based design and optimization of their pharmacological and pharmaceutical agents. Structural studies of potassium channels have revolutionized biophysical understandings of key concepts in the field - ion selectivity, conduction, channel gating, and modulation, making them multi-modality targets of pharmacological regulation. In this chapter, I will select a few high-resolution structures to illustrate key structural insights, proposed allostery behind channel functions, disagreements still open to debate, and channel-lipid interactions and co-evolution. The known structural consensus allows the inference of conserved molecular mechanisms shared among subfamilies of K+ channels and makes it possible to develop channel-specific pharmaceutical agents.
Keywords: Activation, deactivation, and inactivation; Co-evolution of channels and lipids; Energetics and allostery; Ligand-gated K+ channels; Lipid-dependent gating; Pharmacological regulators and small molecule compounds; Structure-based drug design; Voltage-gated K+ channels (Kv).
© 2021. The Author(s), under exclusive license to Springer Nature Switzerland AG.
Conflict of interest statement
Figures


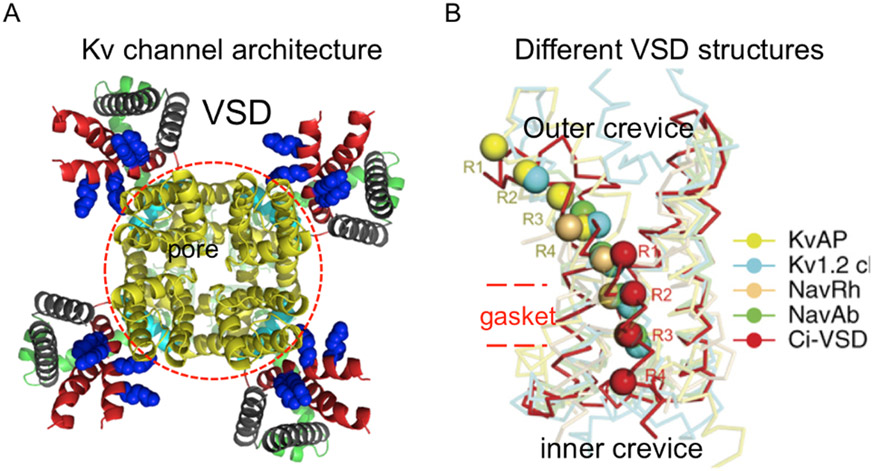
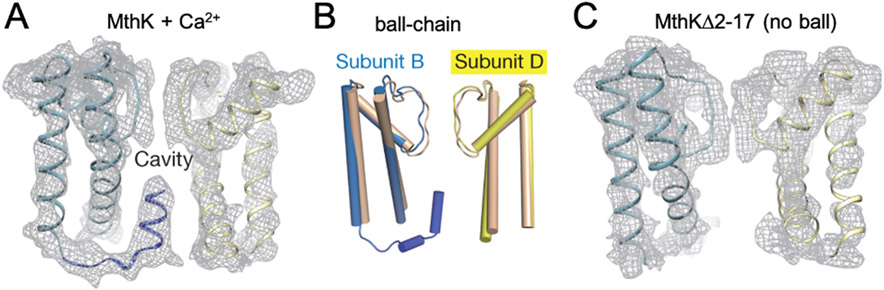





References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
