

The CRTC1-MAML2 fusion is the major oncogenic driver in mucoepidermoid carcinoma
- PMID: 33830080
- PMCID: PMC8119194
- DOI: 10.1172/jci.insight.139497
The CRTC1-MAML2 fusion is the major oncogenic driver in mucoepidermoid carcinoma
Abstract
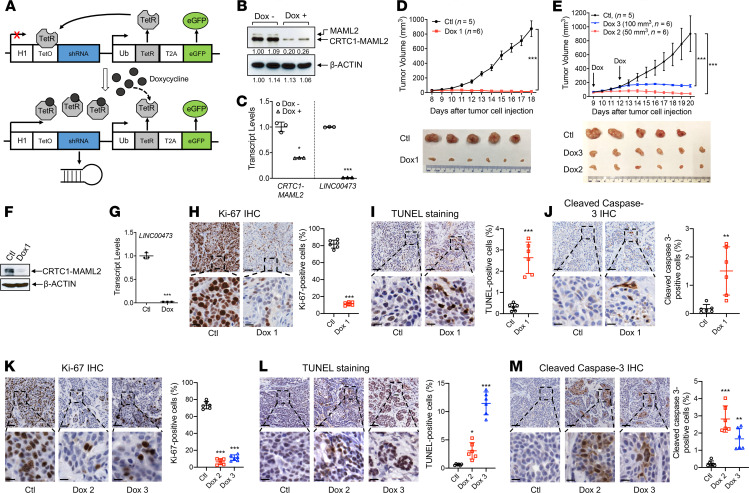
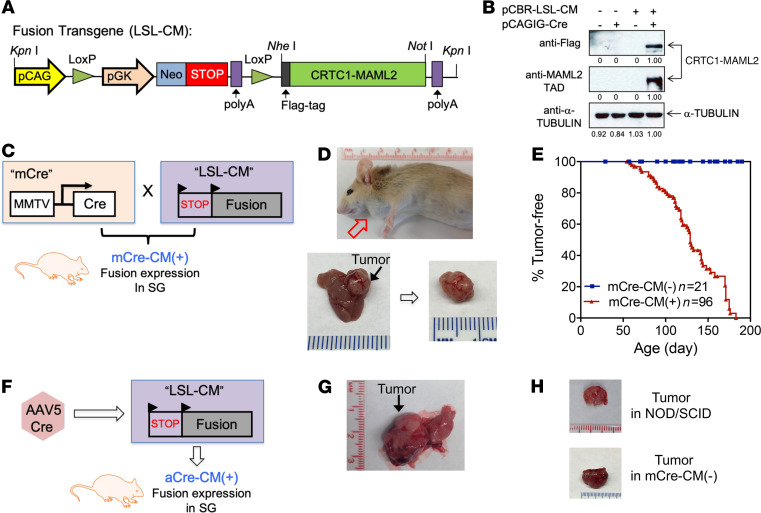
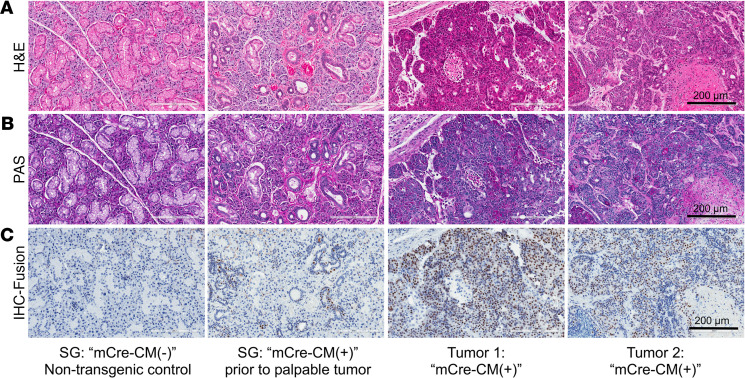
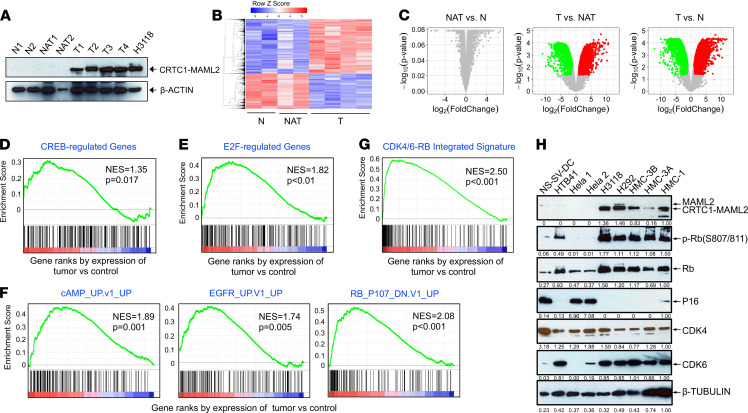
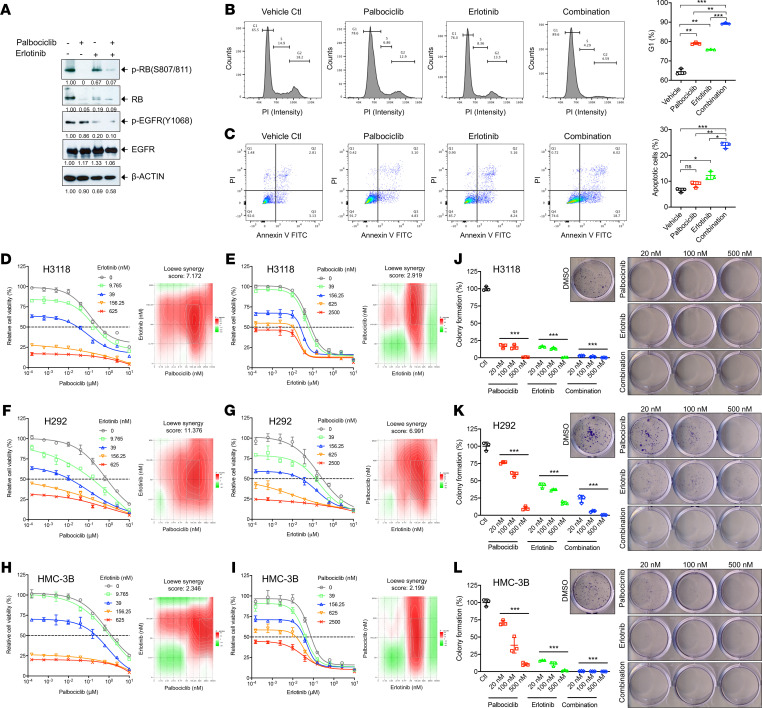
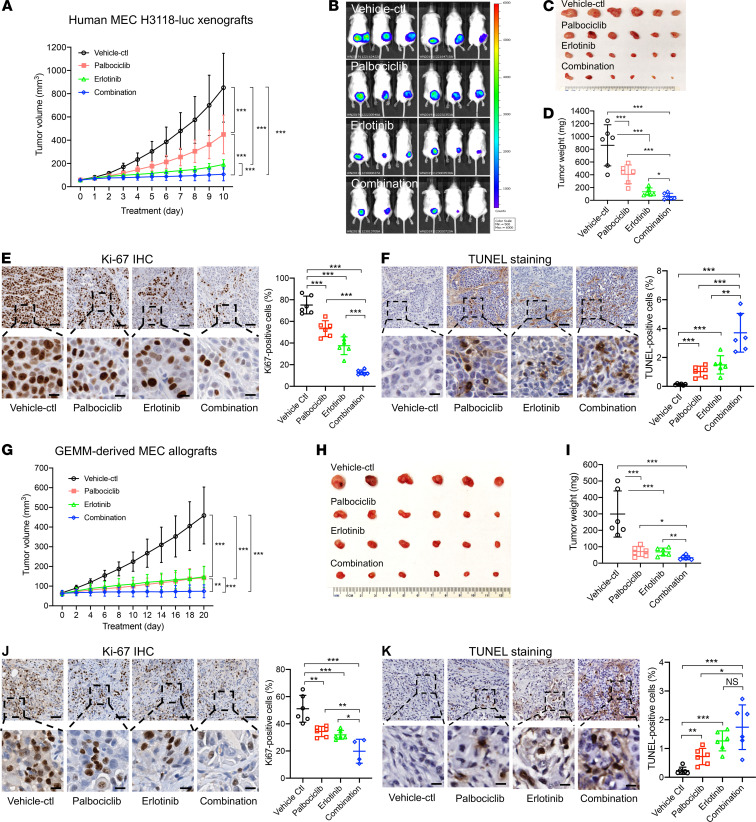
No effective systemic treatment is available for patients with unresectable, recurrent, or metastatic mucoepidermoid carcinoma (MEC), the most common salivary gland malignancy. MEC is frequently associated with a t(11;19)(q14-21;p12-13) translocation that creates a CRTC1-MAML2 fusion gene. The CRTC1-MAML2 fusion exhibited transforming activity in vitro; however, whether it serves as an oncogenic driver for MEC establishment and maintenance in vivo remains unknown. Here, we show that doxycycline-induced CRTC1-MAML2 knockdown blocked the growth of established MEC xenografts, validating CRTC1-MAML2 as a therapeutic target. We further generated a conditional transgenic mouse model and observed that Cre-induced CRTC1-MAML2 expression caused 100% penetrant formation of salivary gland tumors resembling histological and molecular characteristics of human MEC. Molecular analysis of MEC tumors revealed altered p16-CDK4/6-RB pathway activity as a potential cooperating event in promoting CRTC1-MAML2-induced tumorigenesis. Cotargeting of aberrant p16-CDK4/6-RB signaling and CRTC1-MAML2 fusion-activated AREG/EGFR signaling with the respective CDK4/6 inhibitor Palbociclib and EGFR inhibitor Erlotinib produced enhanced antitumor responses in vitro and in vivo. Collectively, this study provides direct evidence for CRTC1-MAML2 as a key driver for MEC development and maintenance and identifies a potentially novel combination therapy with FDA-approved EGFR and CDK4/6 inhibitors as a potential viable strategy for patients with MEC.
Keywords: Head and neck cancer; Mouse models; Oncogenes; Oncology.
Conflict of interest statement
Figures
Similar articles
-
Targeting Notch and EGFR signaling in human mucoepidermoid carcinoma.Signal Transduct Target Ther. 2021 Jan 21;6(1):27. doi: 10.1038/s41392-020-00388-0. Signal Transduct Target Ther. 2021. PMID: 33473104 Free PMC article.
-
Aberrantly activated AREG-EGFR signaling is required for the growth and survival of CRTC1-MAML2 fusion-positive mucoepidermoid carcinoma cells.Oncogene. 2014 Jul 17;33(29):3869-77. doi: 10.1038/onc.2013.348. Epub 2013 Aug 26. Oncogene. 2014. PMID: 23975434
-
CRTC1-MAML2 fusion-induced lncRNA LINC00473 expression maintains the growth and survival of human mucoepidermoid carcinoma cells.Oncogene. 2018 Apr;37(14):1885-1895. doi: 10.1038/s41388-017-0104-0. Epub 2018 Jan 22. Oncogene. 2018. PMID: 29353885 Free PMC article.
-
t(11;19) translocation and CRTC1-MAML2 fusion oncogene in mucoepidermoid carcinoma.Oral Oncol. 2009 Jan;45(1):2-9. doi: 10.1016/j.oraloncology.2008.03.012. Epub 2008 May 16. Oral Oncol. 2009. PMID: 18486532 Review.
-
Prognostic value of CRTC1-MAML2 translocation in salivary mucoepidermoid carcinoma: Systematic review and meta-analysis.J Oral Pathol Med. 2020 May;49(5):386-394. doi: 10.1111/jop.12970. Epub 2019 Nov 16. J Oral Pathol Med. 2020. PMID: 31661572
Cited by
-
Parotid Gland Tumors: Molecular Diagnostic Approaches.Int J Mol Sci. 2024 Jul 4;25(13):7350. doi: 10.3390/ijms25137350. Int J Mol Sci. 2024. PMID: 39000457 Free PMC article. Review.
-
Significance of Notch Signaling in Salivary Gland Development and Diseases.J Clin Med. 2025 May 10;14(10):3325. doi: 10.3390/jcm14103325. J Clin Med. 2025. PMID: 40429321 Free PMC article. Review.
-
An FGFR-p53 developmental signaling axis drives salivary cancer progression.Oncogene. 2025 Aug;44(32):2876-2892. doi: 10.1038/s41388-025-03444-7. Epub 2025 Jun 2. Oncogene. 2025. PMID: 40456865
-
Shining Light on Rarity: Mucoepidermoid Carcinoma of the Esophagus with Absence of MAML2 Gene Rearrangement.Indian J Surg Oncol. 2024 Sep;15(Suppl 3):374-378. doi: 10.1007/s13193-024-01961-7. Epub 2024 May 17. Indian J Surg Oncol. 2024. PMID: 39328726
-
miR-22 and miR-205 Drive Tumor Aggressiveness of Mucoepidermoid Carcinomas of Salivary Glands.Front Oncol. 2022 Feb 9;11:786150. doi: 10.3389/fonc.2021.786150. eCollection 2021. Front Oncol. 2022. PMID: 35223452 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
