

Atractylenolide I enhances responsiveness to immune checkpoint blockade therapy by activating tumor antigen presentation
- PMID: 33830945
- PMCID: PMC8121519
- DOI: 10.1172/JCI146832
Atractylenolide I enhances responsiveness to immune checkpoint blockade therapy by activating tumor antigen presentation
Abstract
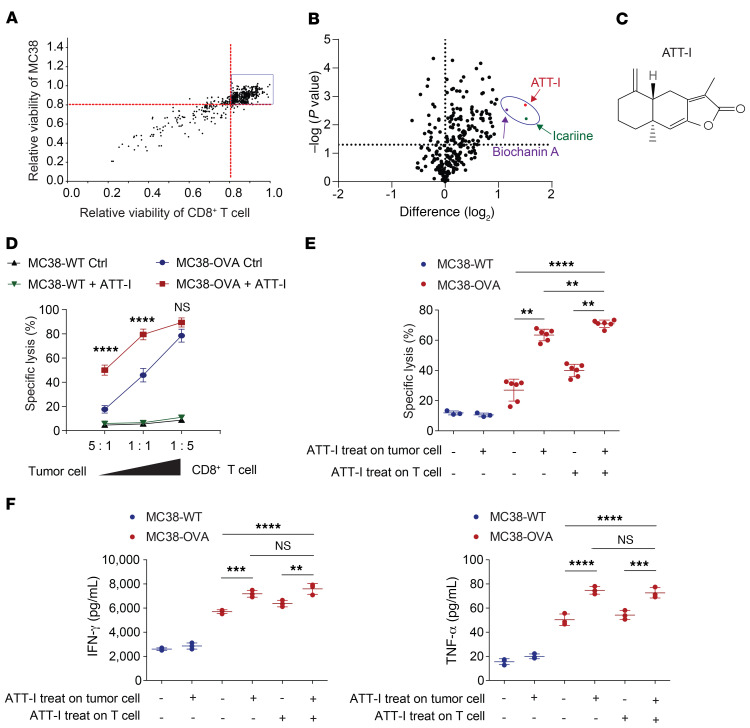
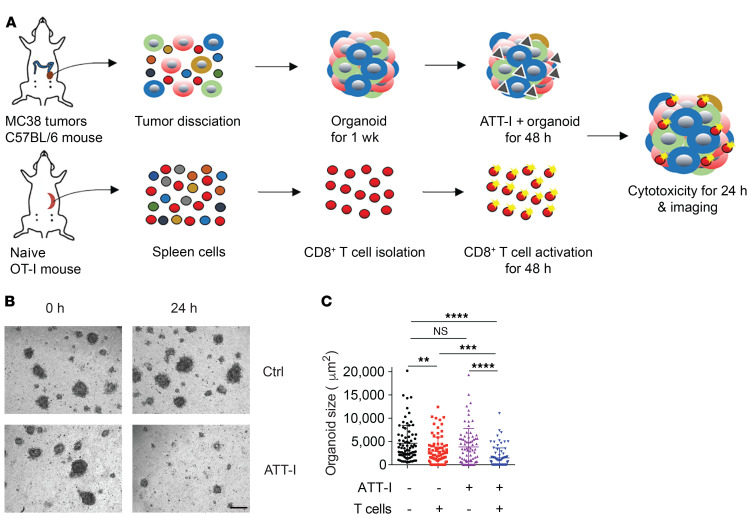
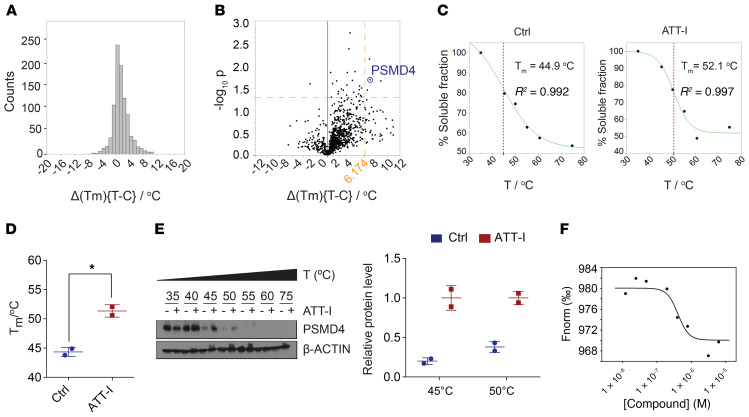
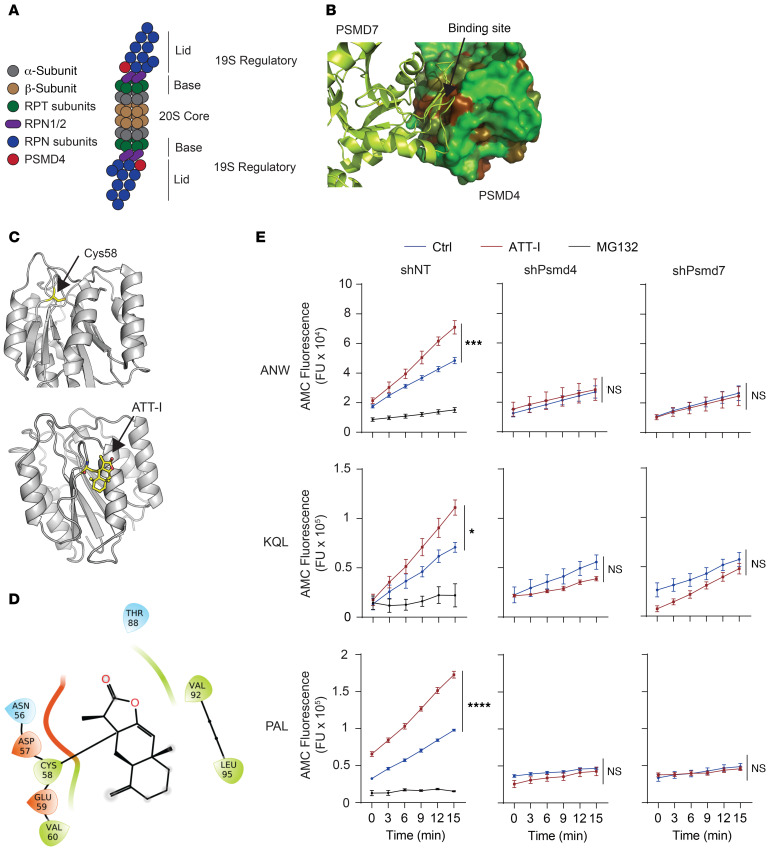
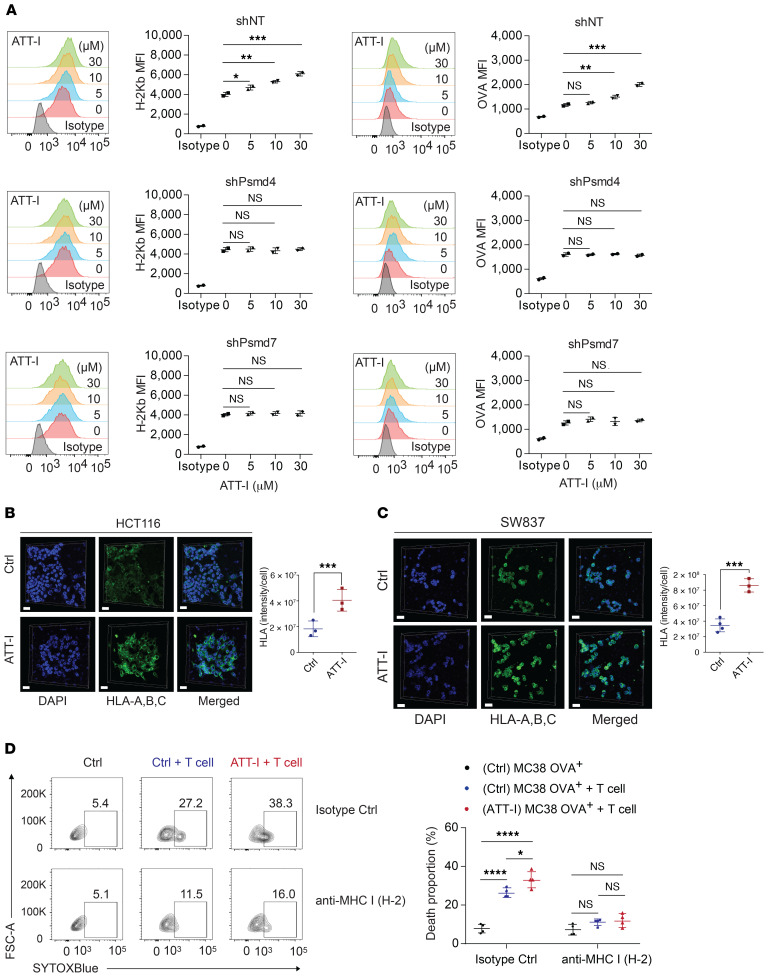
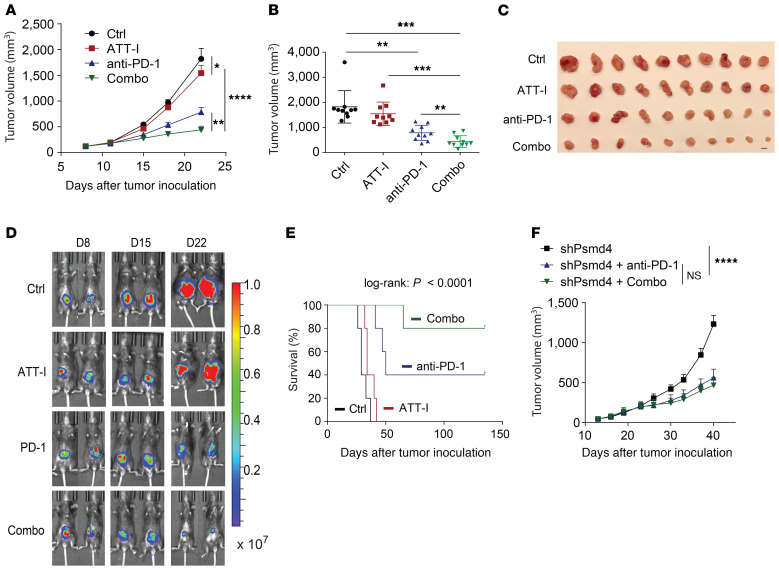
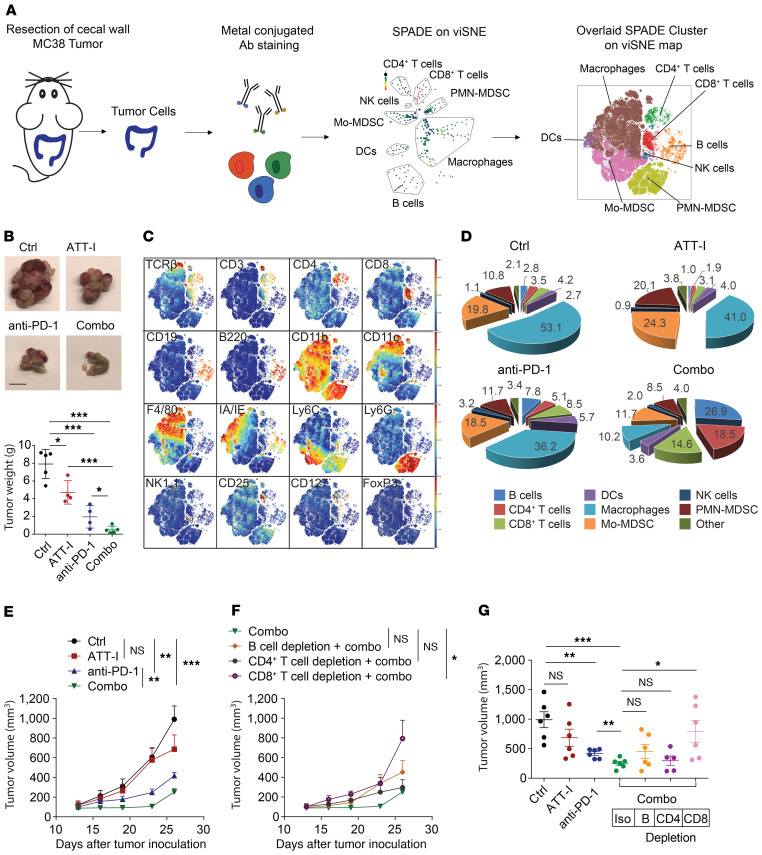
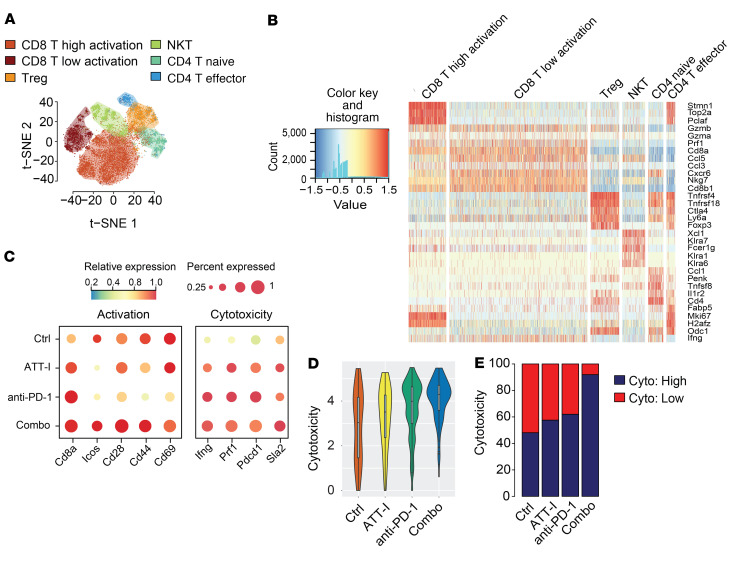
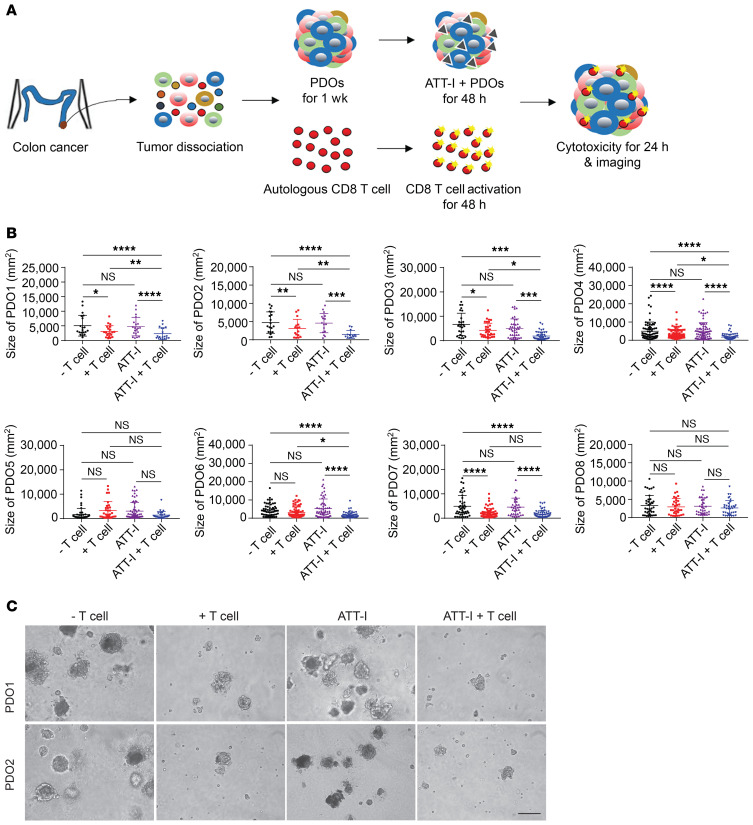
One of the primary mechanisms of tumor cell immune evasion is the loss of antigenicity, which arises due to lack of immunogenic tumor antigens as well as dysregulation of the antigen processing machinery. In a screen for small-molecule compounds from herbal medicine that potentiate T cell-mediated cytotoxicity, we identified atractylenolide I (ATT-I), which substantially promotes tumor antigen presentation of both human and mouse colorectal cancer (CRC) cells and thereby enhances the cytotoxic response of CD8+ T cells. Cellular thermal shift assay (CETSA) with multiplexed quantitative mass spectrometry identified the proteasome 26S subunit non-ATPase 4 (PSMD4), an essential component of the immunoproteasome complex, as a primary target protein of ATT-I. Binding of ATT-I with PSMD4 augments the antigen-processing activity of immunoproteasome, leading to enhanced MHC-I-mediated antigen presentation on cancer cells. In syngeneic mouse CRC models and human patient-derived CRC organoid models, ATT-I treatment promotes the cytotoxicity of CD8+ T cells and thus profoundly enhances the efficacy of immune checkpoint blockade therapy. Collectively, we show here that targeting the function of immunoproteasome with ATT-I promotes tumor antigen presentation and empowers T cell cytotoxicity, thus elevating the tumor response to immunotherapy.
Keywords: Antigen processing; Cancer immunotherapy; Colorectal cancer; Immunology; Oncology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
