

Leveraging expression from multiple tissues using sparse canonical correlation analysis and aggregate tests improves the power of transcriptome-wide association studies
- PMID: 33831007
- PMCID: PMC8057593
- DOI: 10.1371/journal.pgen.1008973
Leveraging expression from multiple tissues using sparse canonical correlation analysis and aggregate tests improves the power of transcriptome-wide association studies
Abstract
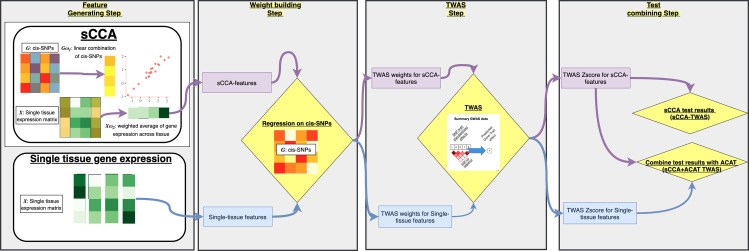
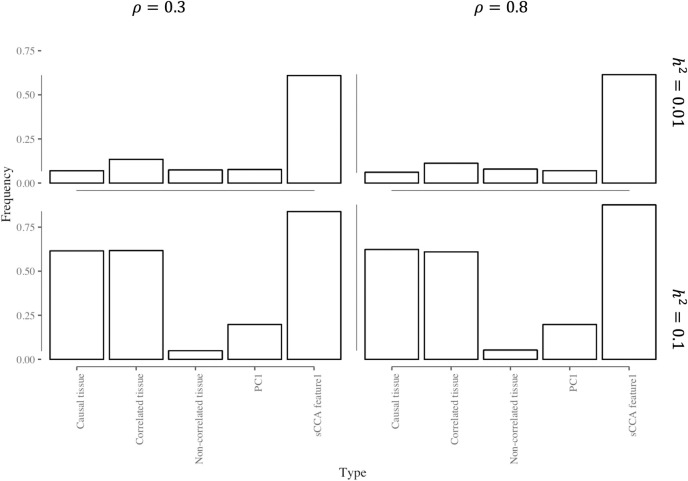
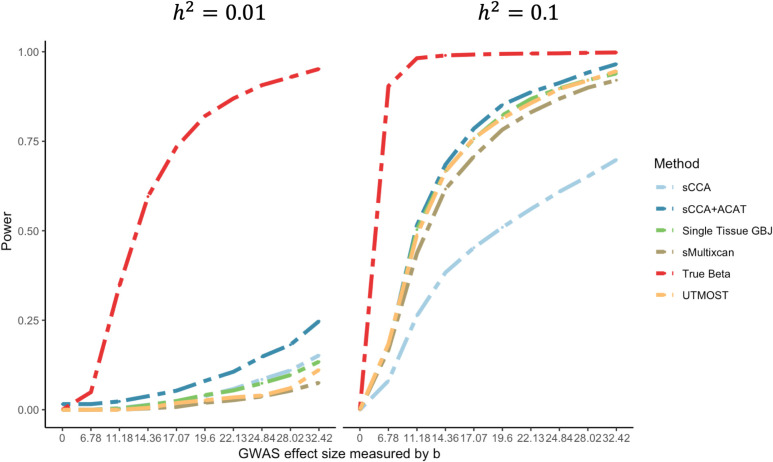
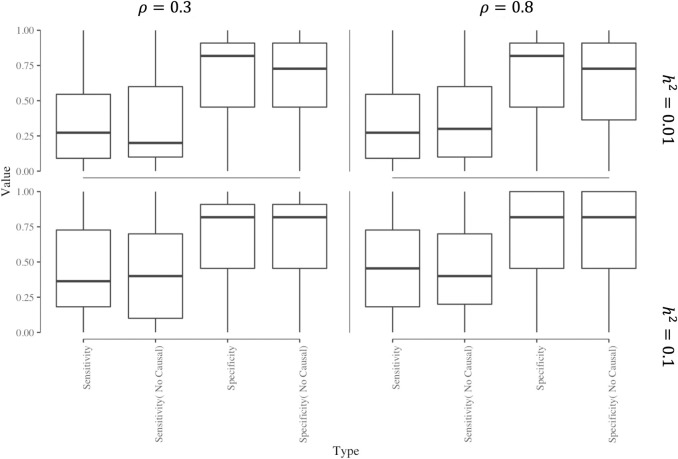
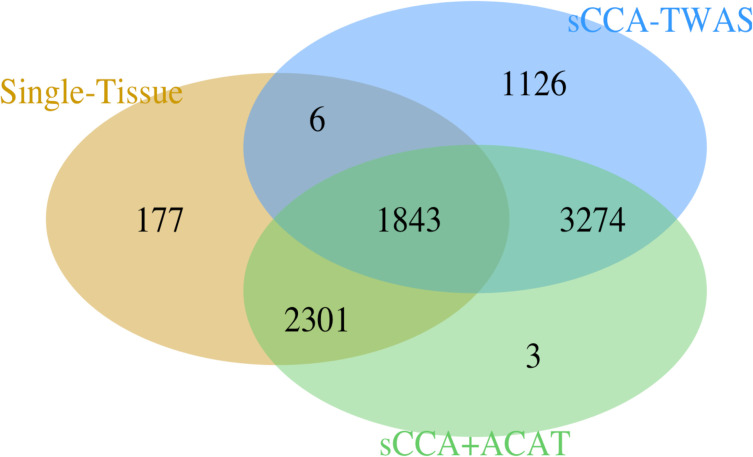
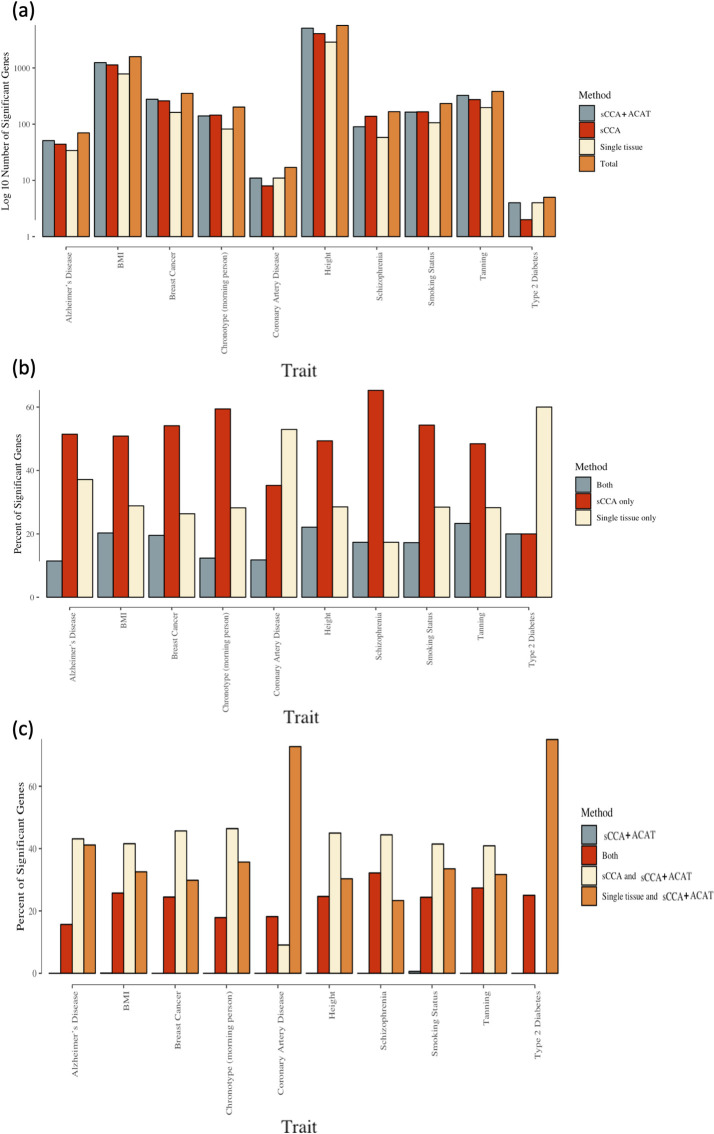
Transcriptome-wide association studies (TWAS) test the association between traits and genetically predicted gene expression levels. The power of a TWAS depends in part on the strength of the correlation between a genetic predictor of gene expression and the causally relevant gene expression values. Consequently, TWAS power can be low when expression quantitative trait locus (eQTL) data used to train the genetic predictors have small sample sizes, or when data from causally relevant tissues are not available. Here, we propose to address these issues by integrating multiple tissues in the TWAS using sparse canonical correlation analysis (sCCA). We show that sCCA-TWAS combined with single-tissue TWAS using an aggregate Cauchy association test (ACAT) outperforms traditional single-tissue TWAS. In empirically motivated simulations, the sCCA+ACAT approach yielded the highest power to detect a gene associated with phenotype, even when expression in the causal tissue was not directly measured, while controlling the Type I error when there is no association between gene expression and phenotype. For example, when gene expression explains 2% of the variability in outcome, and the GWAS sample size is 20,000, the average power difference between the ACAT combined test of sCCA features and single-tissue, versus single-tissue combined with Generalized Berk-Jones (GBJ) method, single-tissue combined with S-MultiXcan, UTMOST, or summarizing cross-tissue expression patterns using Principal Component Analysis (PCA) approaches was 5%, 8%, 5% and 38%, respectively. The gain in power is likely due to sCCA cross-tissue features being more likely to be detectably heritable. When applied to publicly available summary statistics from 10 complex traits, the sCCA+ACAT test was able to increase the number of testable genes and identify on average an additional 400 additional gene-trait associations that single-trait TWAS missed. Our results suggest that aggregating eQTL data across multiple tissues using sCCA can improve the sensitivity of TWAS while controlling for the false positive rate.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
