

High-throughput, single-copy sequencing reveals SARS-CoV-2 spike variants coincident with mounting humoral immunity during acute COVID-19
- PMID: 33831133
- PMCID: PMC8031304
- DOI: 10.1371/journal.ppat.1009431
High-throughput, single-copy sequencing reveals SARS-CoV-2 spike variants coincident with mounting humoral immunity during acute COVID-19
Abstract
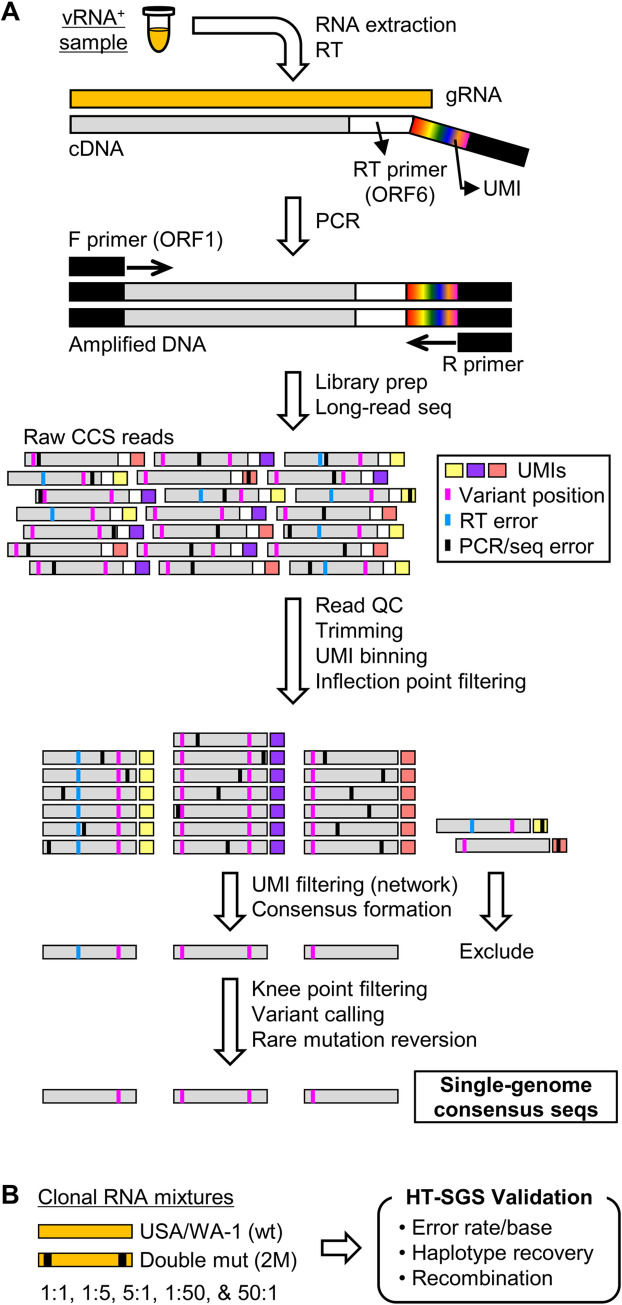
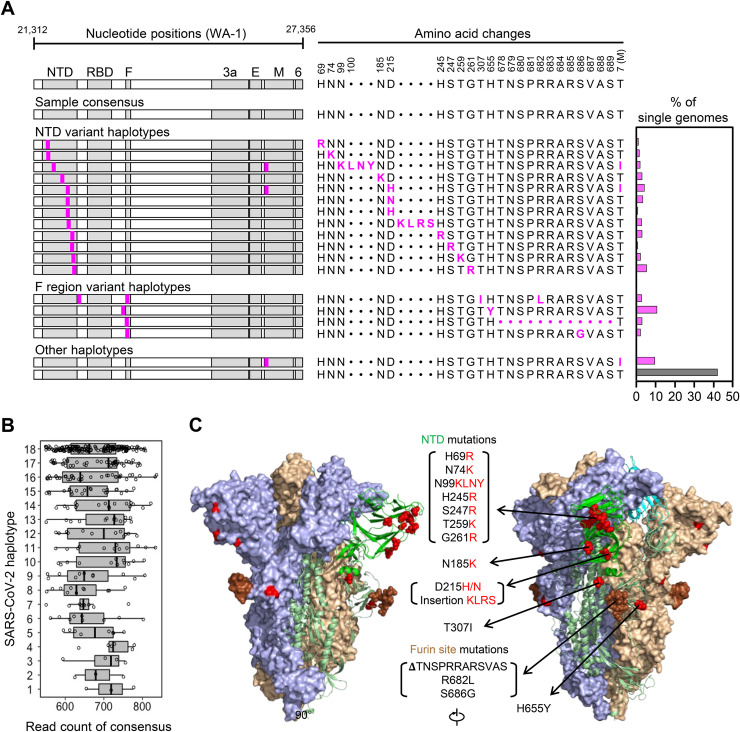
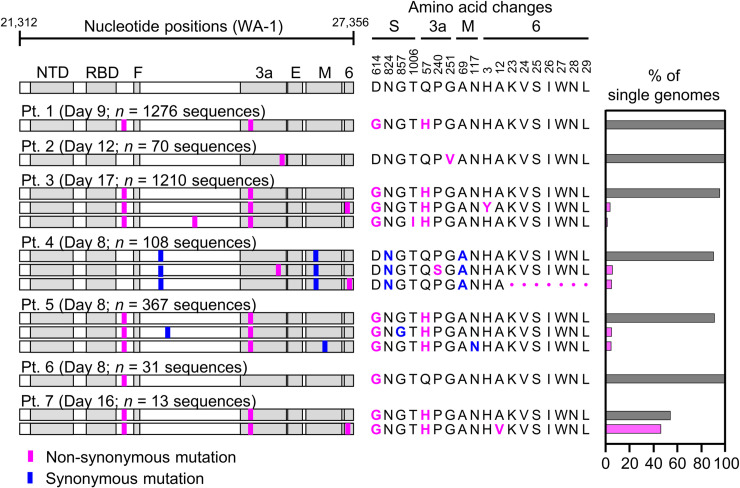
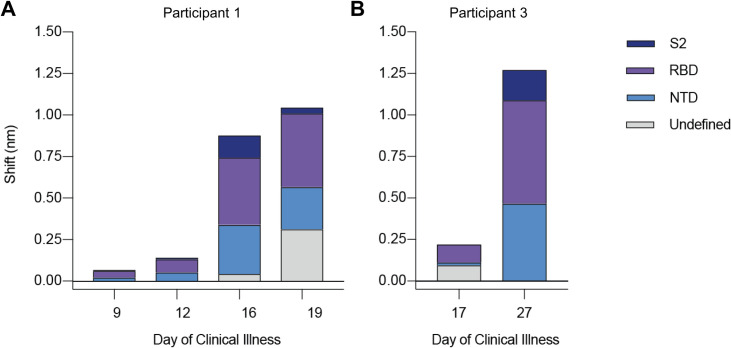
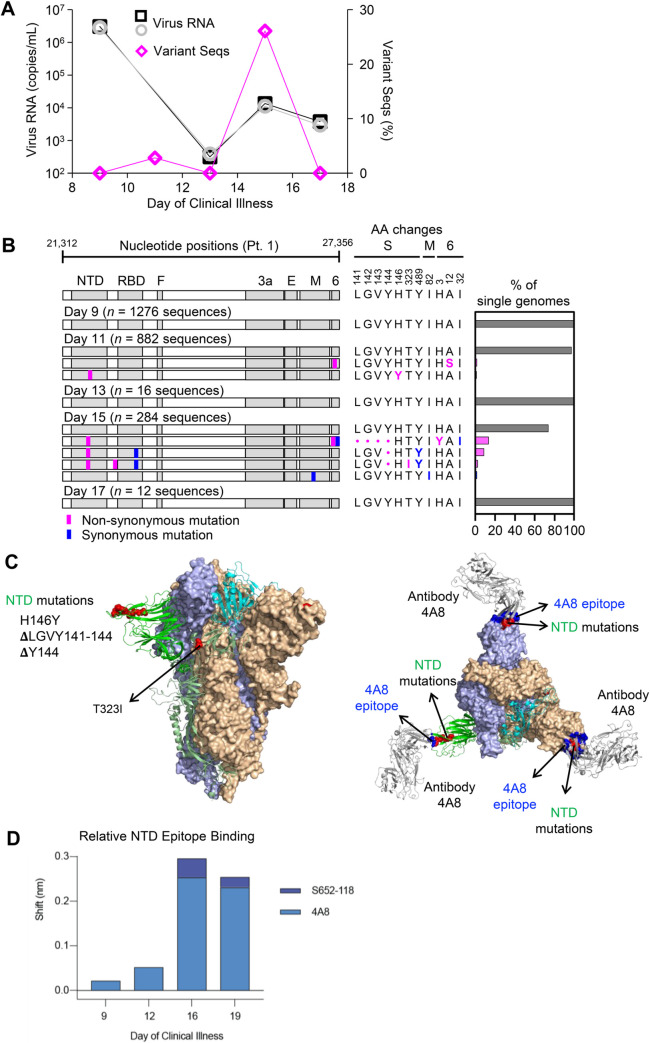
Tracking evolution of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) within infected individuals will help elucidate coronavirus disease 2019 (COVID-19) pathogenesis and inform use of antiviral interventions. In this study, we developed an approach for sequencing the region encoding the SARS-CoV-2 virion surface proteins from large numbers of individual virus RNA genomes per sample. We applied this approach to the WA-1 reference clinical isolate of SARS-CoV-2 passaged in vitro and to upper respiratory samples from 7 study participants with COVID-19. SARS-CoV-2 genomes from cell culture were diverse, including 18 haplotypes with non-synonymous mutations clustered in the spike NH2-terminal domain (NTD) and furin cleavage site regions. By contrast, cross-sectional analysis of samples from participants with COVID-19 showed fewer virus variants, without structural clustering of mutations. However, longitudinal analysis in one individual revealed 4 virus haplotypes bearing 3 independent mutations in a spike NTD epitope targeted by autologous antibodies. These mutations arose coincident with a 6.2-fold rise in serum binding to spike and a transient increase in virus burden. We conclude that SARS-CoV-2 exhibits a capacity for rapid genetic adaptation that becomes detectable in vivo with the onset of humoral immunity, with the potential to contribute to delayed virologic clearance in the acute setting.
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
High-Throughput, Single-Copy Sequencing Reveals SARS-CoV-2 Spike Variants Coincident with Mounting Humoral Immunity during Acute COVID-19.bioRxiv [Preprint]. 2021 Feb 22:2021.02.21.432184. doi: 10.1101/2021.02.21.432184. bioRxiv. 2021. Update in: PLoS Pathog. 2021 Apr 8;17(4):e1009431. doi: 10.1371/journal.ppat.1009431. PMID: 33655255 Free PMC article. Updated. Preprint.
References
-
- To KK-W, Tsang OT-Y, Leung W-S, Tam AR, Wu T-C, Lung DC, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. The Lancet Infectious Diseases. 2020;20(5):565–74. 10.1016/S1473-3099(20)30196-1 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
