

Allele-specific expression of GATA2 due to epigenetic dysregulation in CEBPA double-mutant AML
- PMID: 33831168
- PMCID: PMC8489178
- DOI: 10.1182/blood.2020009244
Allele-specific expression of GATA2 due to epigenetic dysregulation in CEBPA double-mutant AML
Erratum in
-
Mulet-Lazaro R, van Herk S, Erpelinck C, et al. Allele-specific expression of GATA2 due to epigenetic dysregulation in CEBPA double-mutant AML. Blood. 2021;138(2):160-177.Blood. 2022 Mar 17;139(11):1772. doi: 10.1182/blood.2022015740. Blood. 2022. PMID: 35298597 Free PMC article. No abstract available.
Abstract
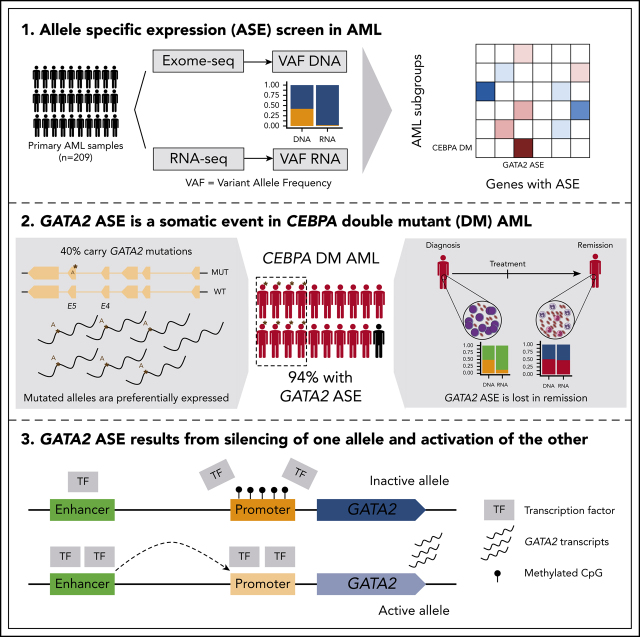
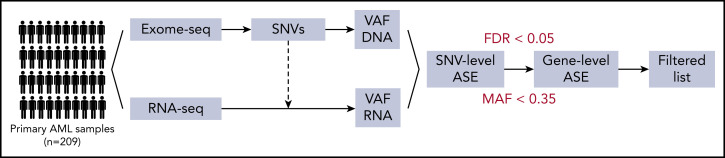
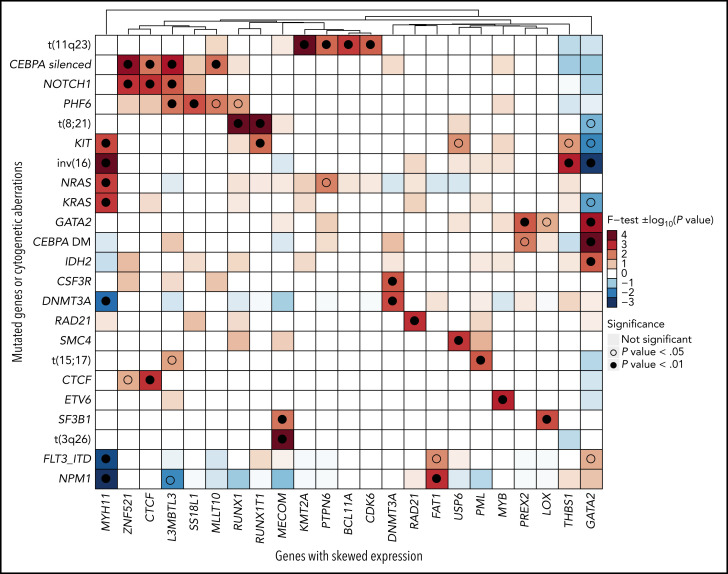
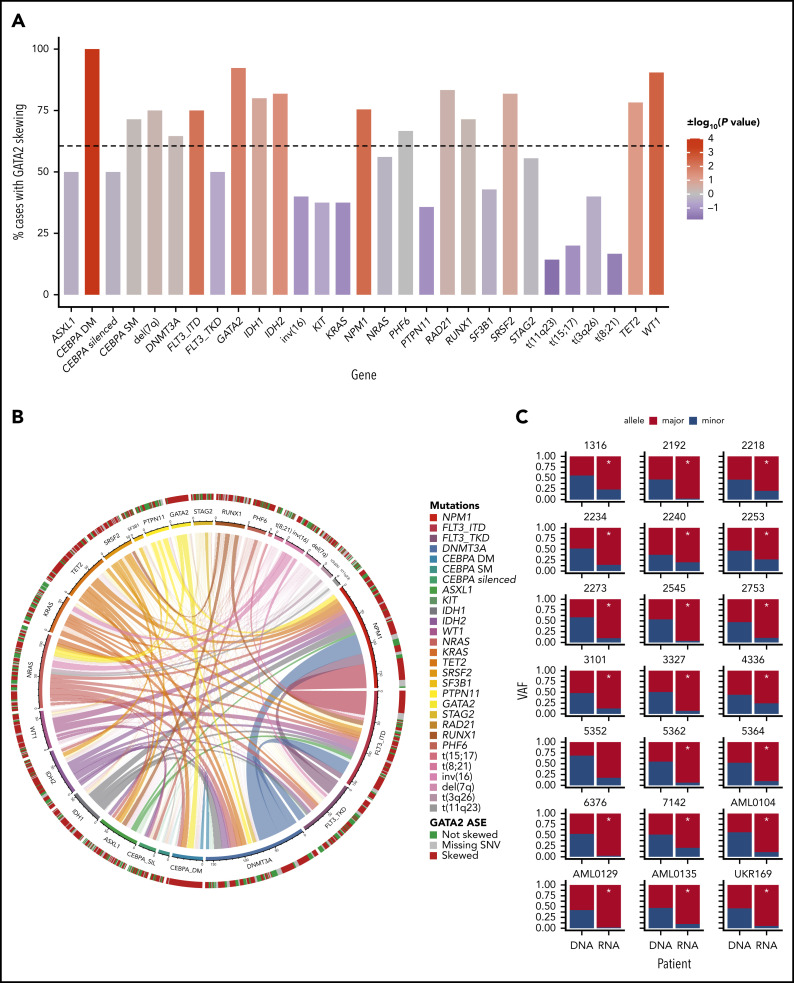
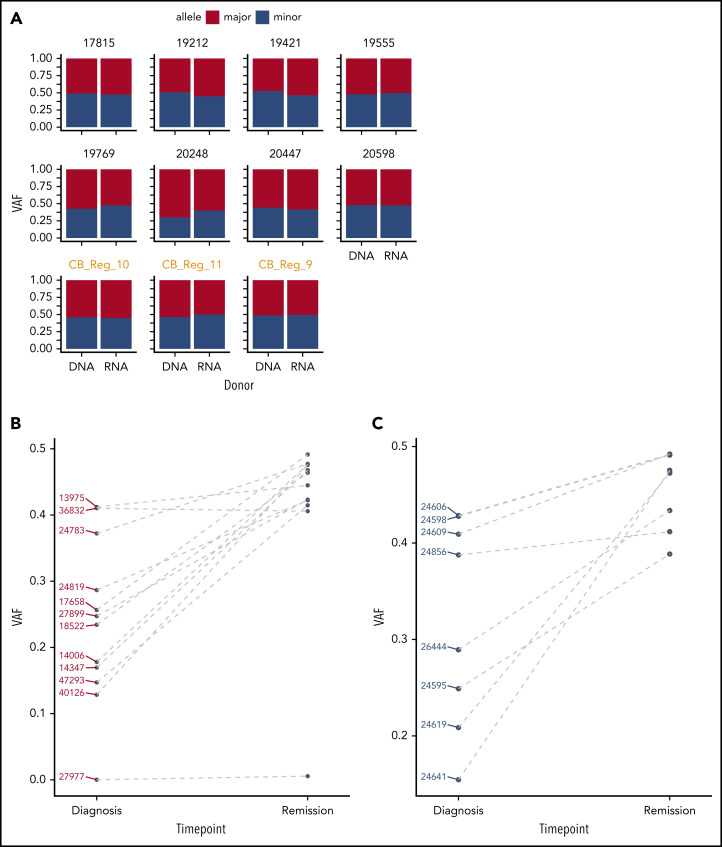
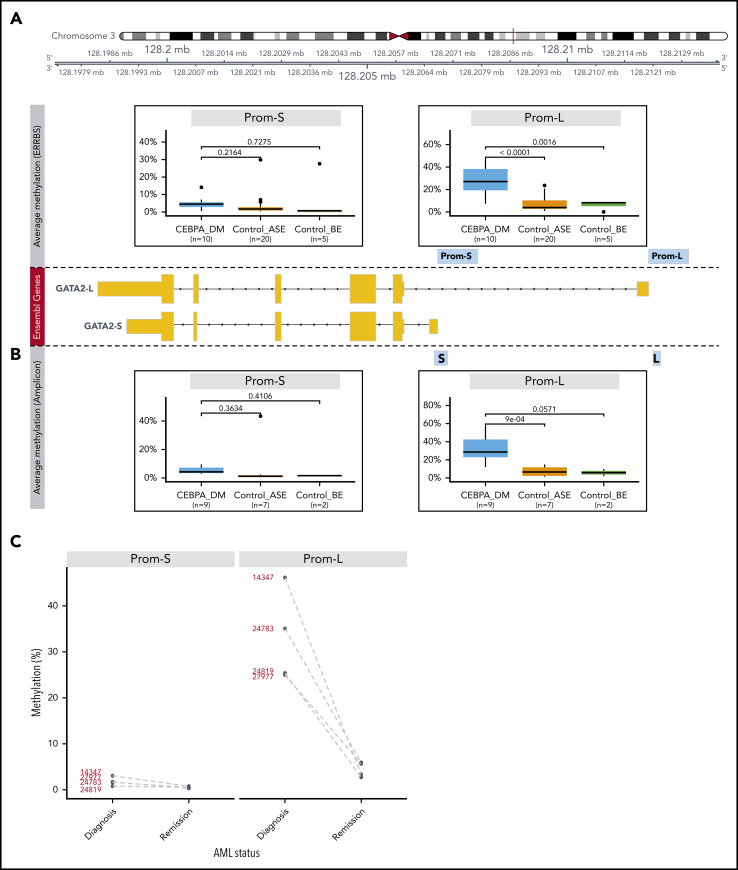
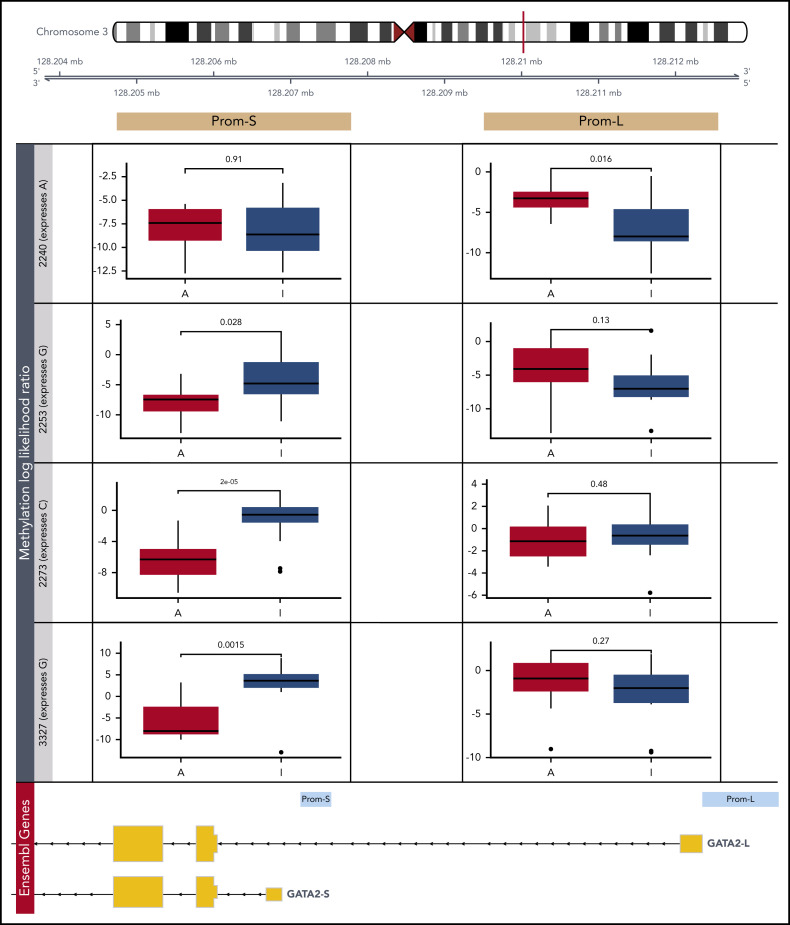
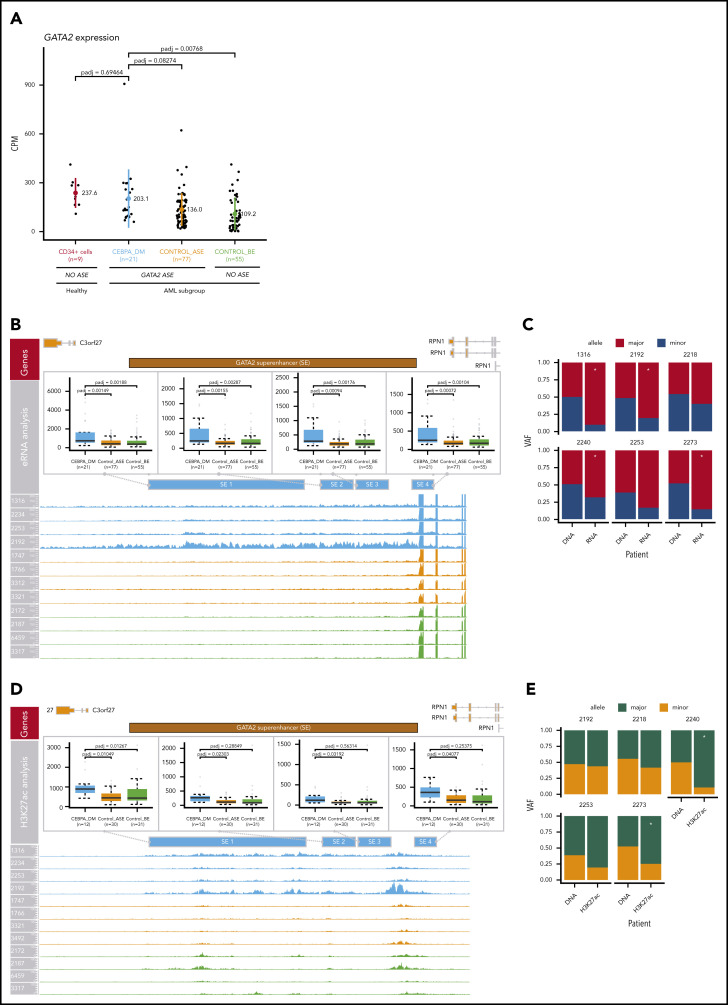
Transcriptional deregulation is a central event in the development of acute myeloid leukemia (AML). To identify potential disturbances in gene regulation, we conducted an unbiased screen of allele-specific expression (ASE) in 209 AML cases. The gene encoding GATA binding protein 2 (GATA2) displayed ASE more often than any other myeloid- or cancer-related gene. GATA2 ASE was strongly associated with CEBPA double mutations (DMs), with 95% of cases presenting GATA2 ASE. In CEBPA DM AML with GATA2 mutations, the mutated allele was preferentially expressed. We found that GATA2 ASE was a somatic event lost in complete remission, supporting the notion that it plays a role in CEBPA DM AML. Acquisition of GATA2 ASE involved silencing of 1 allele via promoter methylation and concurrent overactivation of the other allele, thereby preserving expression levels. Notably, promoter methylation was also lost in remission along with GATA2 ASE. In summary, we propose that GATA2 ASE is acquired by epigenetic mechanisms and is a prerequisite for the development of AML with CEBPA DMs. This finding constitutes a novel example of an epigenetic hit cooperating with a genetic hit in the pathogenesis of AML.
© 2021 by The American Society of Hematology.
Figures
References
-
- Gröschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157(2):369-381. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
