

Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes
- PMID: 33831375
- PMCID: PMC8054914
- DOI: 10.1016/j.cell.2021.03.009
Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes
Abstract
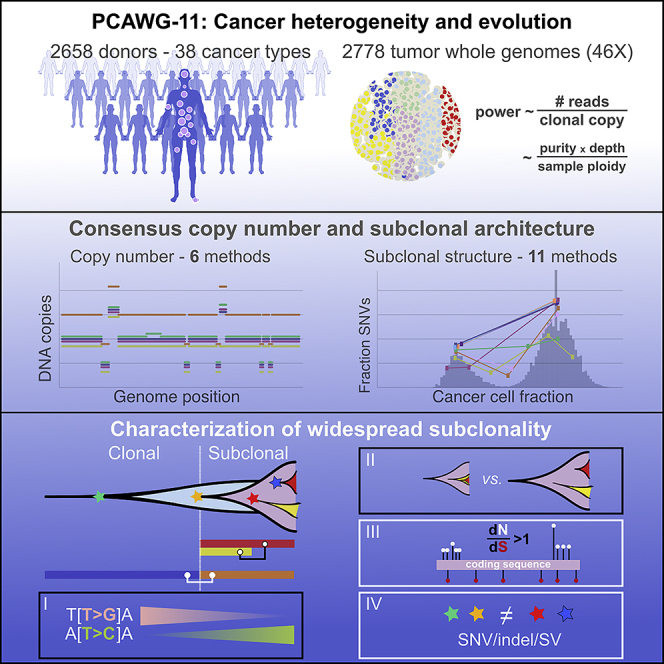
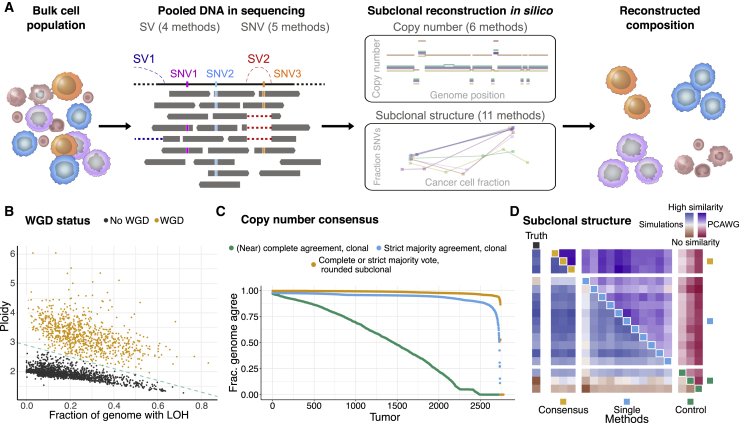
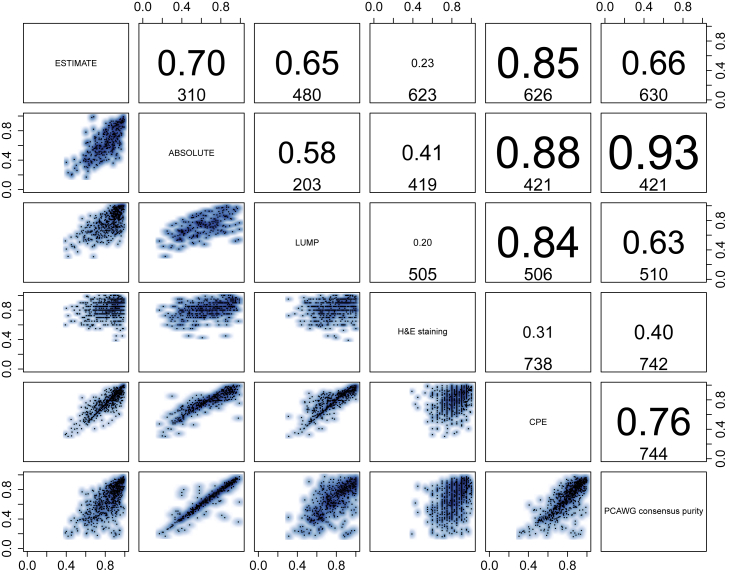
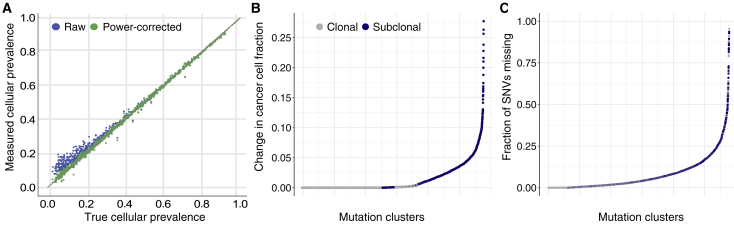
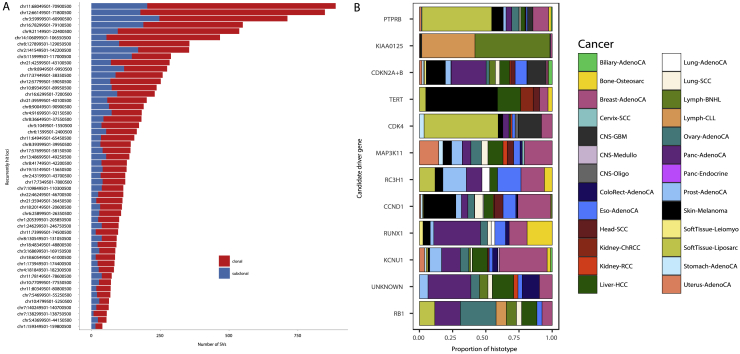
Intra-tumor heterogeneity (ITH) is a mechanism of therapeutic resistance and therefore an important clinical challenge. However, the extent, origin, and drivers of ITH across cancer types are poorly understood. To address this, we extensively characterize ITH across whole-genome sequences of 2,658 cancer samples spanning 38 cancer types. Nearly all informative samples (95.1%) contain evidence of distinct subclonal expansions with frequent branching relationships between subclones. We observe positive selection of subclonal driver mutations across most cancer types and identify cancer type-specific subclonal patterns of driver gene mutations, fusions, structural variants, and copy number alterations as well as dynamic changes in mutational processes between subclonal expansions. Our results underline the importance of ITH and its drivers in tumor evolution and provide a pan-cancer resource of comprehensively annotated subclonal events from whole-genome sequencing data.
Keywords: branching evolution; cancer driver genes; cancer evolution; intra-tumor heterogeneity; pan-cancer genomics; subclonal reconstruction; tumor phylogeny; whole-genome sequencing.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests G.M. and F.M. are cofounders and shareholders of Tailor Bio. R.B. owns equity in Ampressa Therapeutics. G.G. receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MuTect, ABSOLUTE, MutSig, MSMuTect, and POLYSOLVER. I.L. is a consultant for PACT Pharma. B.J.R. is a consultant at and has ownership interest (including stock and patents) in Medley Genomics. N.M. has stock options in and has consulted for Achilles Therapeutics. C.S. acknowledges grant support from Pfizer, AstraZeneca, Bristol Myers Squibb, Roche-Ventana, Boehringer-Ingelheim, Archer Dx, and Ono Pharmaceutical; is an AstraZeneca Advisory Board Member and Chief Investigator for the MeRmaiD-1 clinical trial; has consulted for Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol Myers Squibb, Celgene, AstraZeneca, Illumina, Amgen, Genentech, Roche-Ventana, GRAIL, Medicxi, Bicycle Therapeutics, and the Sarah Cannon Research Institute; has stock options in Apogen Biotechnologies, Epic Bioscience, and GRAIL; and has stock options and is co-founder of Achilles Therapeutics.
Figures
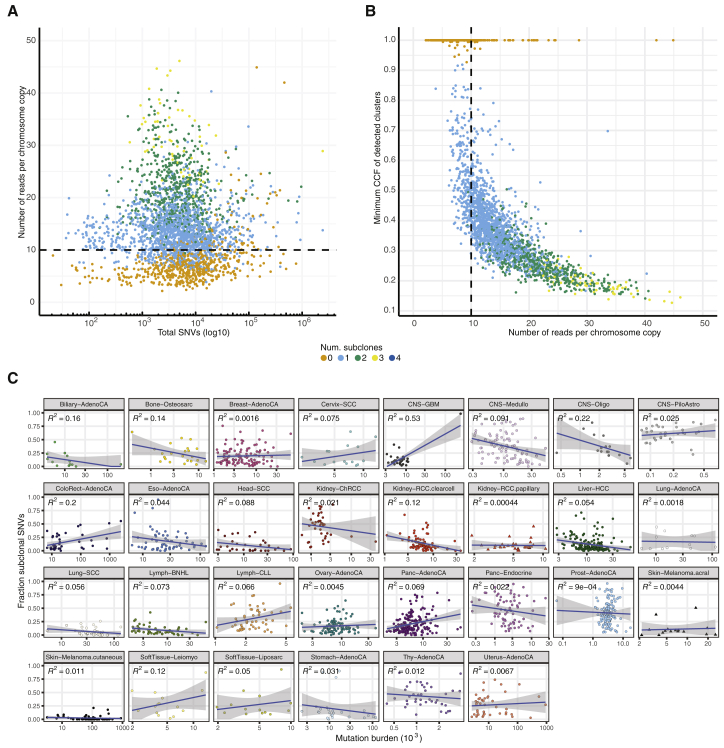
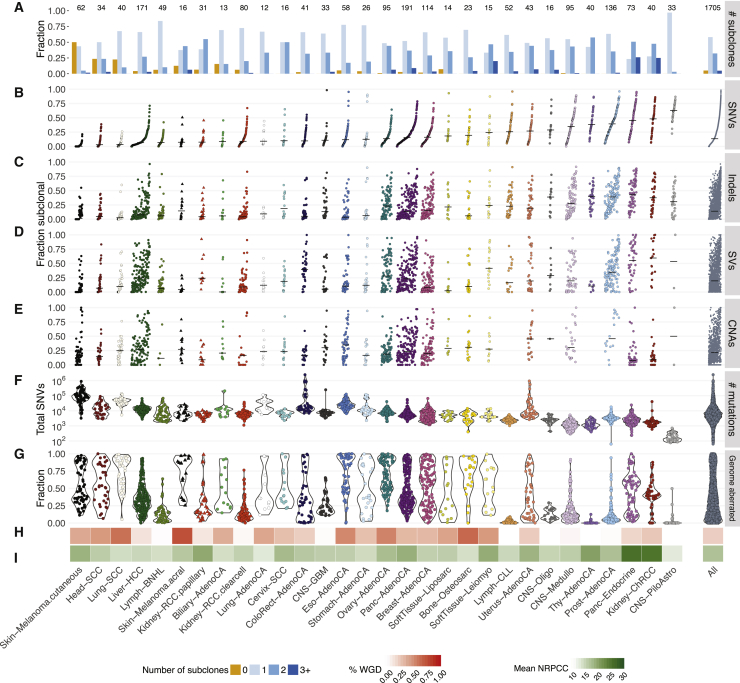
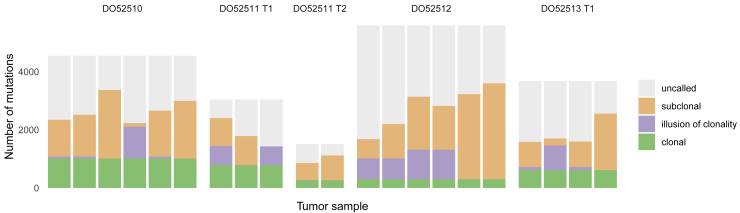
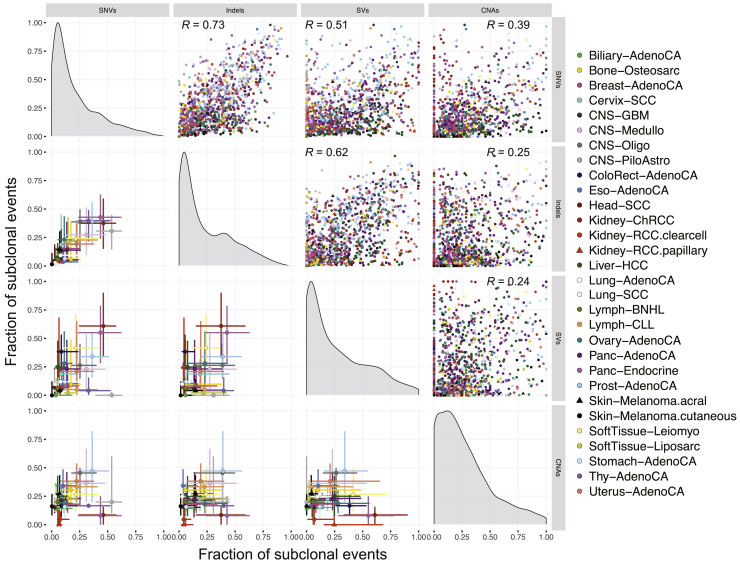
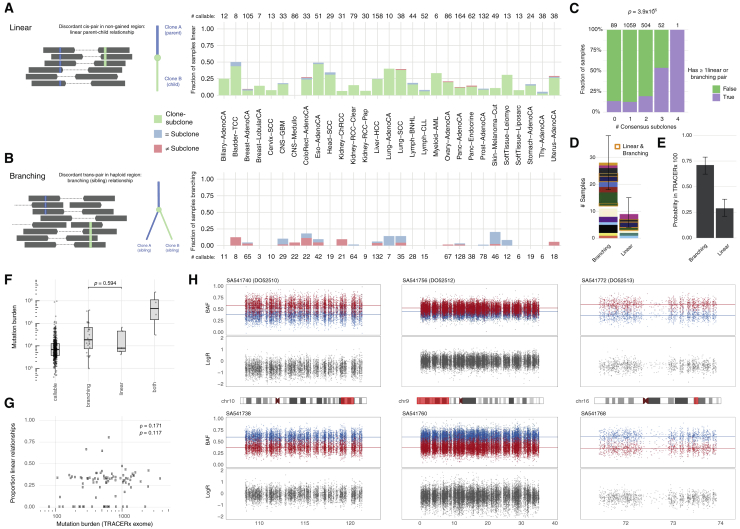
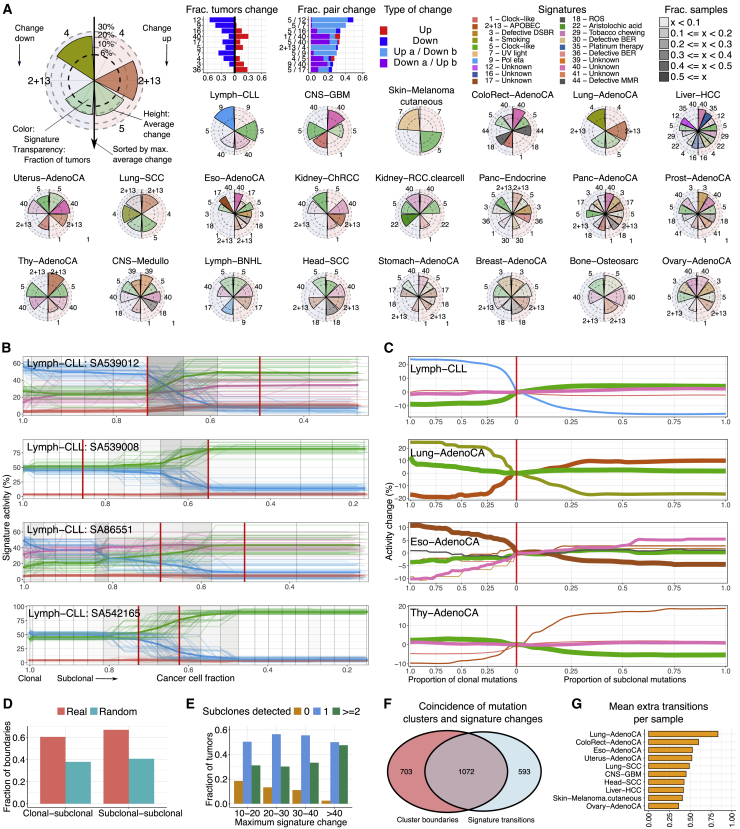
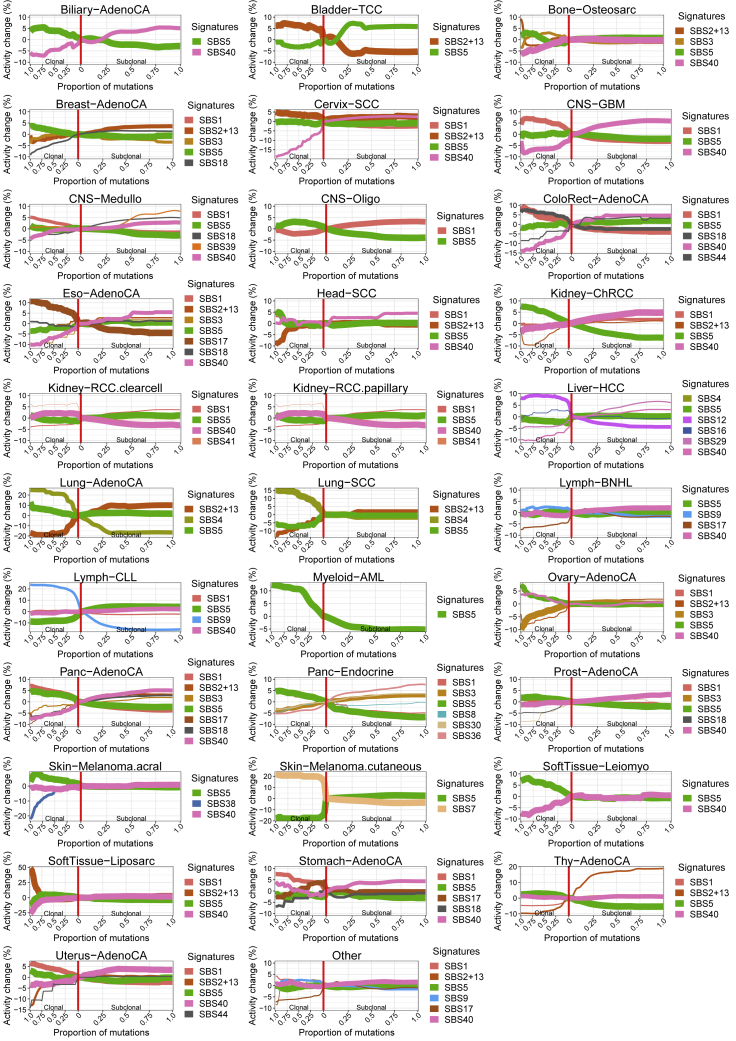
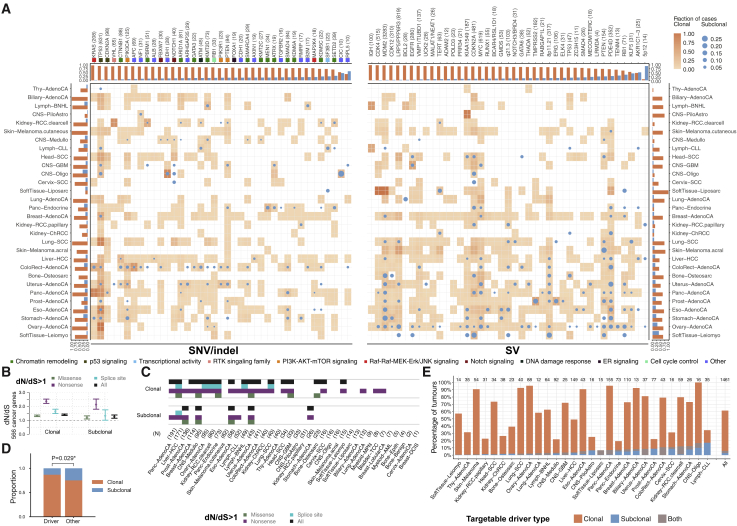
Comment in
-
A large-scale snapshot of intratumor heterogeneity in human cancer.Cancer Cell. 2021 Apr 12;39(4):463-465. doi: 10.1016/j.ccell.2021.03.005. Cancer Cell. 2021. PMID: 33848477
References
-
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 211179/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- FC001202/WT_/Wellcome Trust/United Kingdom
- P30 CA016672/CA/NCI NIH HHS/United States
- MR/V000292/1/MRC_/Medical Research Council/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- U24 CA143799/CA/NCI NIH HHS/United States
- FC001202 /MRC_/Medical Research Council/United Kingdom
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 CA183793/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- FC001202 /CRUK_/Cancer Research UK/United Kingdom
- WT097678/WT_/Wellcome Trust/United Kingdom
- 21777/CRUK_/Cancer Research UK/United Kingdom
- FC001169 /CRUK_/Cancer Research UK/United Kingdom
- FC001169 /MRC_/Medical Research Council/United Kingdom
- R01 CA239342/CA/NCI NIH HHS/United States
- MR/L016311/1/MRC_/Medical Research Council/United Kingdom
- 27176/CRUK_/Cancer Research UK/United Kingdom
- FC001169 /WT_/Wellcome Trust/United Kingdom
- P50 CA211015/CA/NCI NIH HHS/United States
- 21717/CRUK_/Cancer Research UK/United Kingdom
- FC001169/WT_/Wellcome Trust/United Kingdom
- U24 CA210999/CA/NCI NIH HHS/United States
- R01 CA132897/CA/NCI NIH HHS/United States
- FC001202 /WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
