

Interpretation of allele-specific chromatin accessibility using cell state-aware deep learning
- PMID: 33832990
- PMCID: PMC8168584
- DOI: 10.1101/gr.260851.120
Interpretation of allele-specific chromatin accessibility using cell state-aware deep learning
Abstract
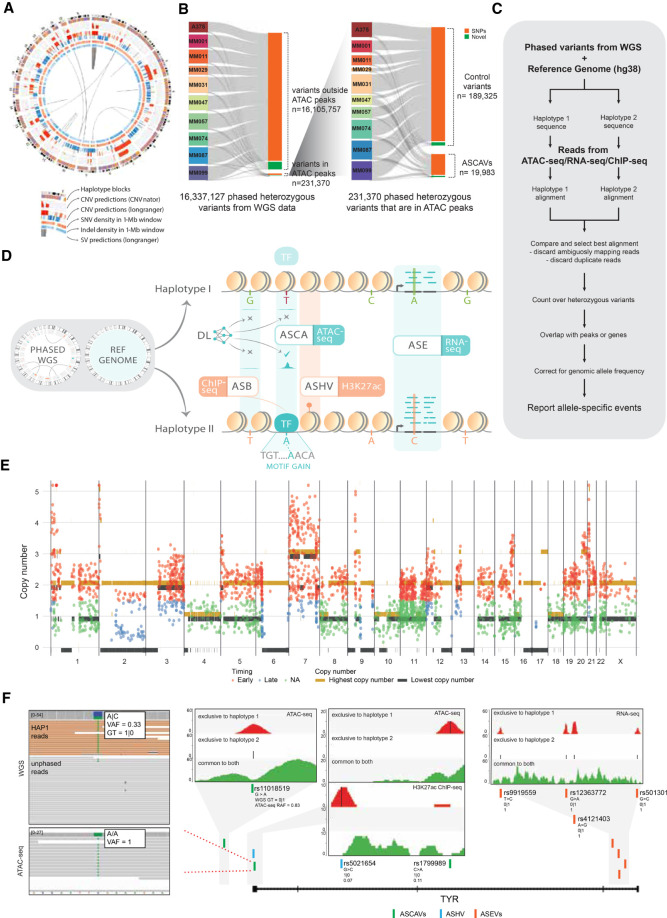
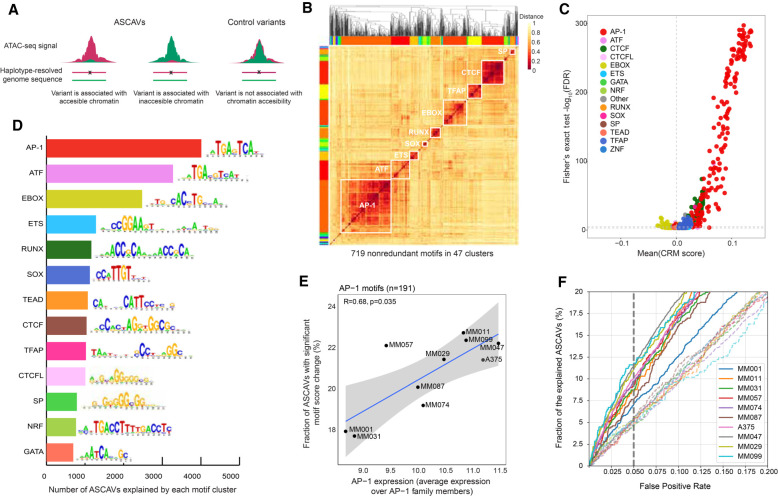
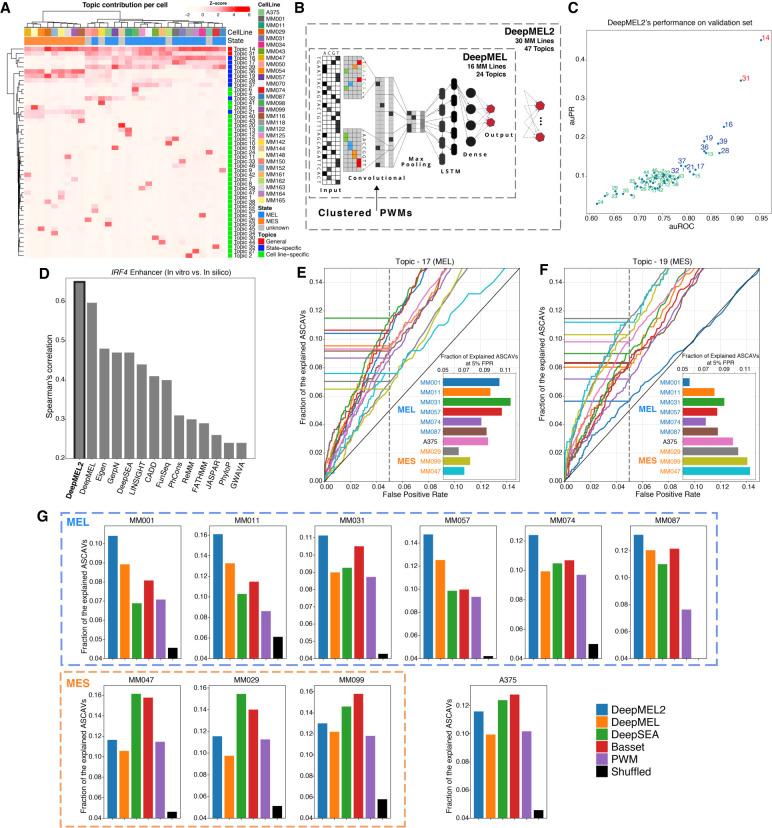
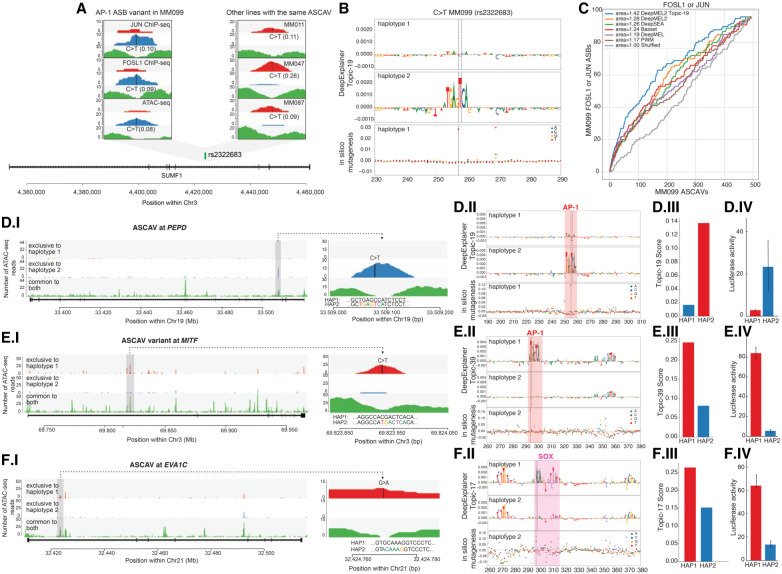
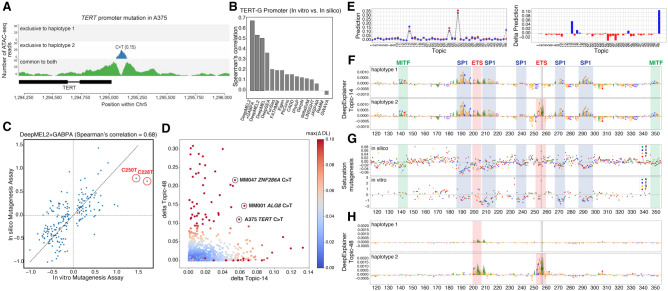
Genomic sequence variation within enhancers and promoters can have a significant impact on the cellular state and phenotype. However, sifting through the millions of candidate variants in a personal genome or a cancer genome, to identify those that impact cis-regulatory function, remains a major challenge. Interpretation of noncoding genome variation benefits from explainable artificial intelligence to predict and interpret the impact of a mutation on gene regulation. Here we generate phased whole genomes with matched chromatin accessibility, histone modifications, and gene expression for 10 melanoma cell lines. We find that training a specialized deep learning model, called DeepMEL2, on melanoma chromatin accessibility data can capture the various regulatory programs of the melanocytic and mesenchymal-like melanoma cell states. This model outperforms motif-based variant scoring, as well as more generic deep learning models. We detect hundreds to thousands of allele-specific chromatin accessibility variants (ASCAVs) in each melanoma genome, of which 15%-20% can be explained by gains or losses of transcription factor binding sites. A considerable fraction of ASCAVs are caused by changes in AP-1 binding, as confirmed by matched ChIP-seq data to identify allele-specific binding of JUN and FOSL1. Finally, by augmenting the DeepMEL2 model with ChIP-seq data for GABPA, the TERT promoter mutation, as well as additional ETS motif gains, can be identified with high confidence. In conclusion, we present a new integrative genomics approach and a deep learning model to identify and interpret functional enhancer mutations with allelic imbalance of chromatin accessibility and gene expression.
© 2021 Atak et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous