

Temporal multiomic modeling reveals a B-cell receptor proliferative program in chronic lymphocytic leukemia
- PMID: 33833385
- PMCID: PMC8102193
- DOI: 10.1038/s41375-021-01221-5
Temporal multiomic modeling reveals a B-cell receptor proliferative program in chronic lymphocytic leukemia
Abstract
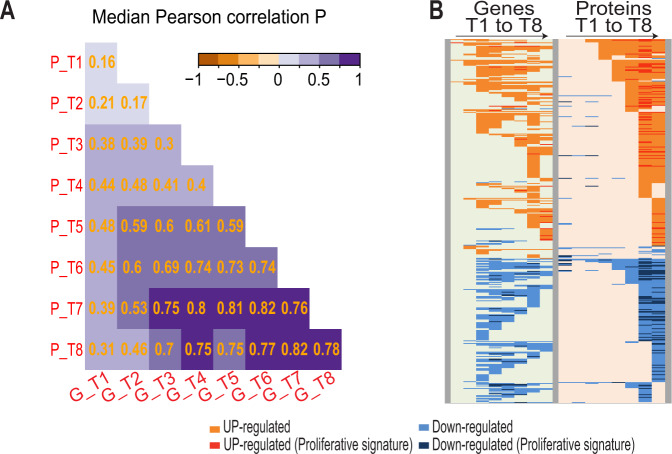
B-cell receptor (BCR) signaling is crucial for the pathophysiology of most mature B-cell lymphomas/leukemias and has emerged as a therapeutic target whose effectiveness remains limited by the occurrence of mutations. Therefore, deciphering the cellular program activated downstream this pathway has become of paramount importance for the development of innovative therapies. Using an original ex vivo model of BCR-induced proliferation of chronic lymphocytic leukemia cells, we generated 108 temporal transcriptional and proteomic profiles from 1 h up to 4 days after BCR activation. This dataset revealed a structured temporal response composed of 13,065 transcripts and 4027 proteins, comprising a leukemic proliferative signature consisting of 430 genes and 374 proteins. Mathematical modeling of this complex cellular response further highlighted a transcriptional network driven by 14 early genes linked to proteins involved in cell proliferation. This group includes expected genes (EGR1/2, NF-kB) and genes involved in NF-kB signaling modulation (TANK, ROHF) and immune evasion (KMO, IL4I1) that have not yet been associated with leukemic cells proliferation. Our study unveils the BCR-activated proliferative genetic program in primary leukemic cells. This approach combining temporal measurements with modeling allows identifying new putative targets for innovative therapy of lymphoid malignancies and also cancers dependent on ligand-receptor interactions.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
