

Advances and opportunities in malaria population genomics
- PMID: 33833443
- PMCID: PMC8028584
- DOI: 10.1038/s41576-021-00349-5
Advances and opportunities in malaria population genomics
Abstract
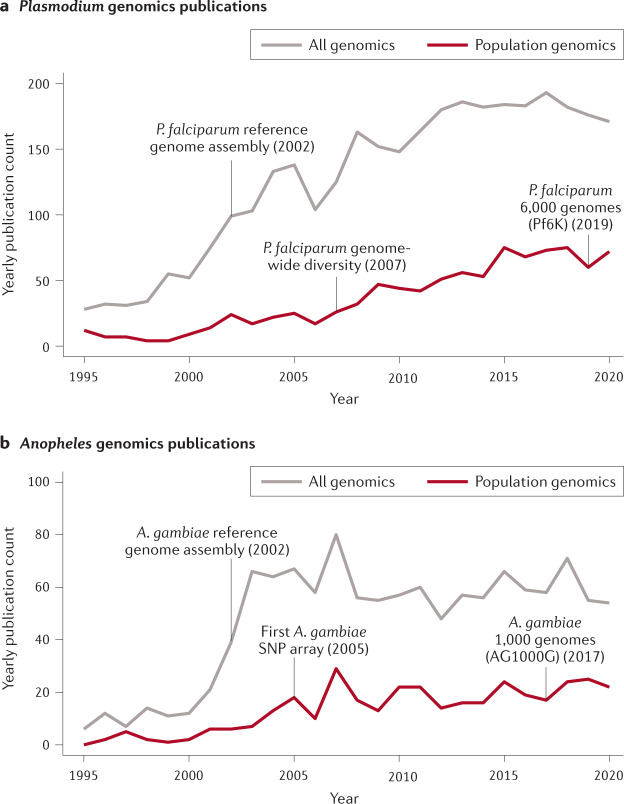
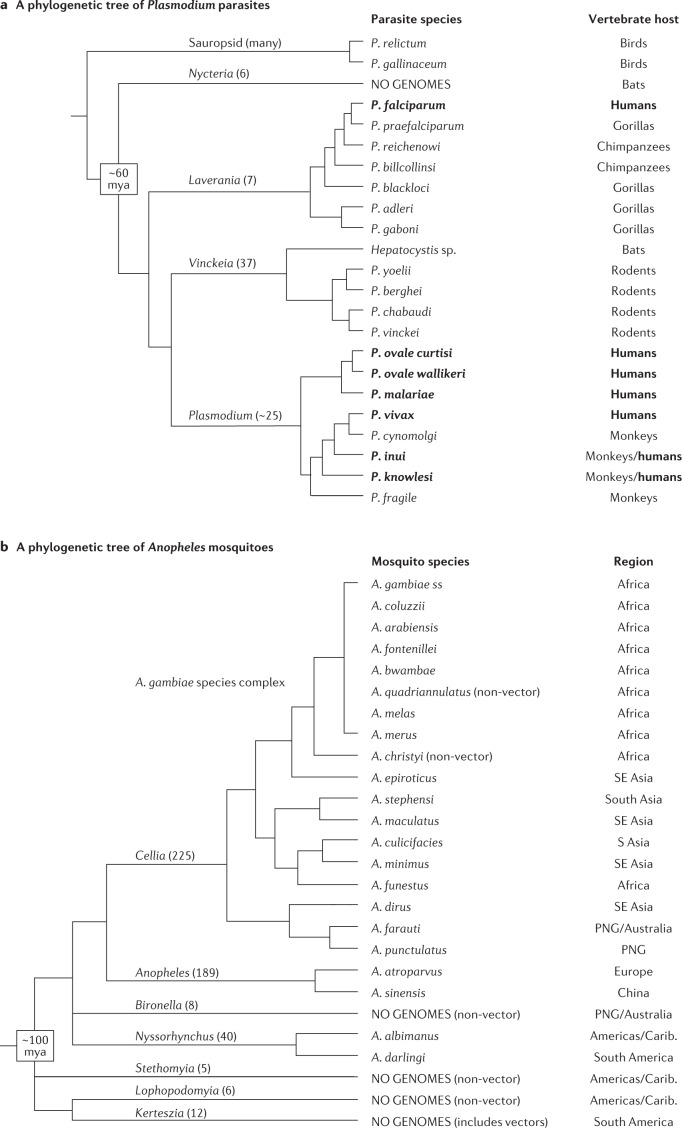
Almost 20 years have passed since the first reference genome assemblies were published for Plasmodium falciparum, the deadliest malaria parasite, and Anopheles gambiae, the most important mosquito vector of malaria in sub-Saharan Africa. Reference genomes now exist for all human malaria parasites and nearly half of the ~40 important vectors around the world. As a foundation for genetic diversity studies, these reference genomes have helped advance our understanding of basic disease biology and drug and insecticide resistance, and have informed vaccine development efforts. Population genomic data are increasingly being used to guide our understanding of malaria epidemiology, for example by assessing connectivity between populations and the efficacy of parasite and vector interventions. The potential value of these applications to malaria control strategies, together with the increasing diversity of genomic data types and contexts in which data are being generated, raise both opportunities and challenges in the field. This Review discusses advances in malaria genomics and explores how population genomic data could be harnessed to further support global disease control efforts.
© 2021. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- World Health Organization. World Malaria Report 2020 (WHO, 2020).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
