

Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA
- PMID: 33833465
- PMCID: PMC9304985
- DOI: 10.1038/s42255-021-00378-8
Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA
Abstract
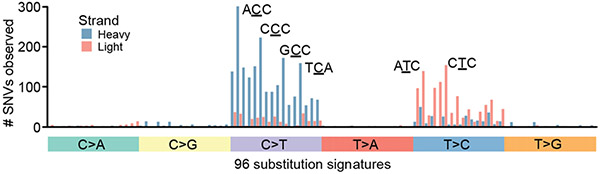
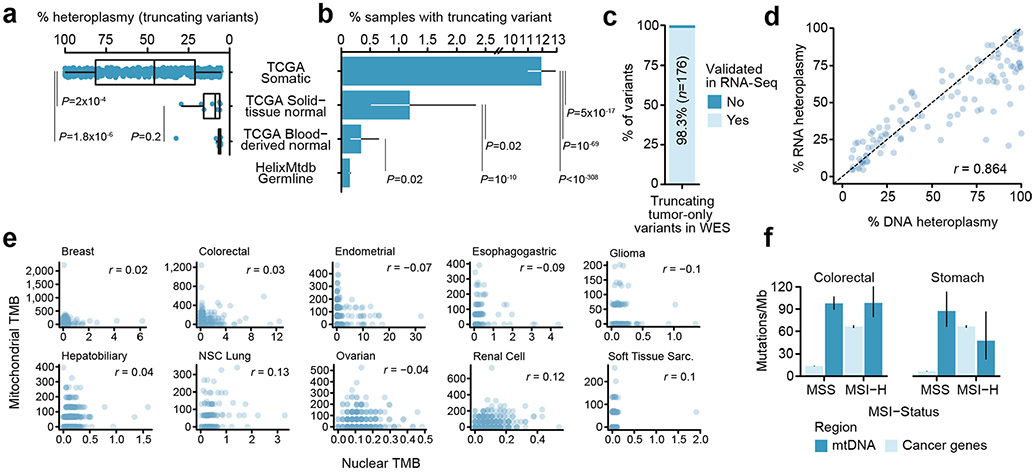
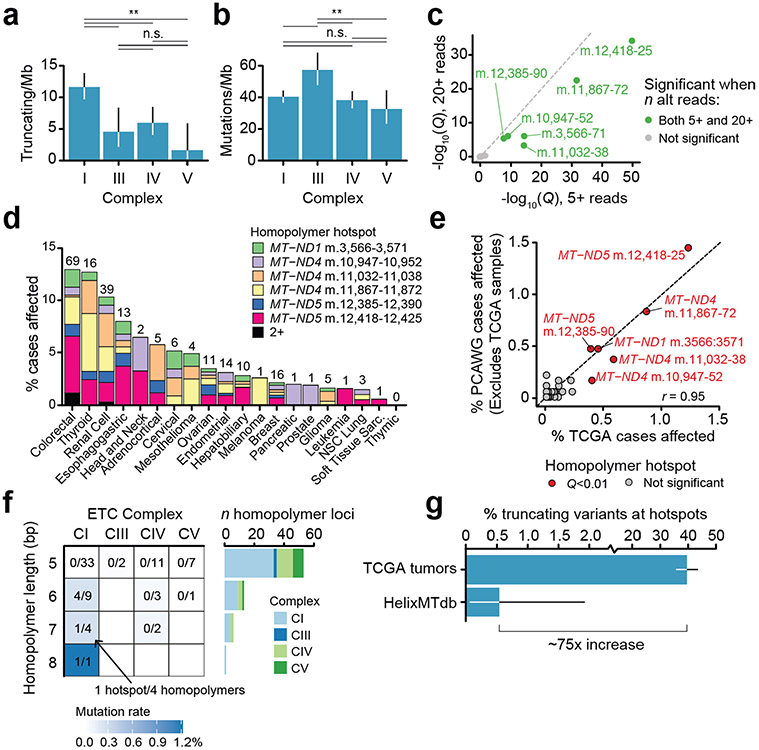
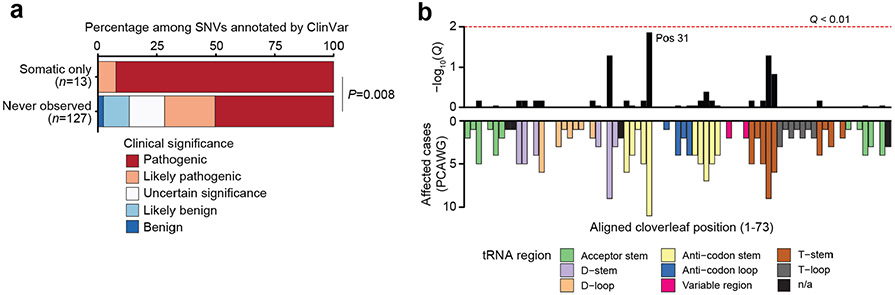
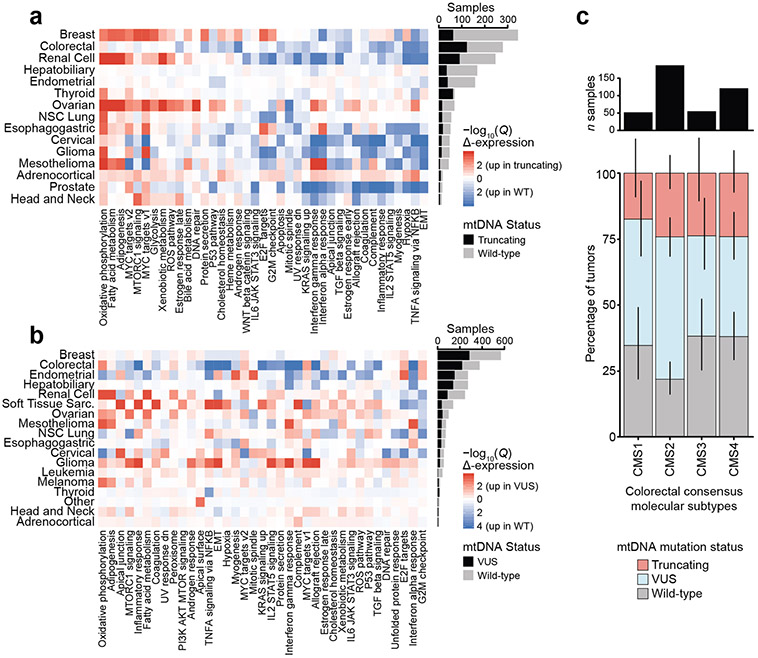
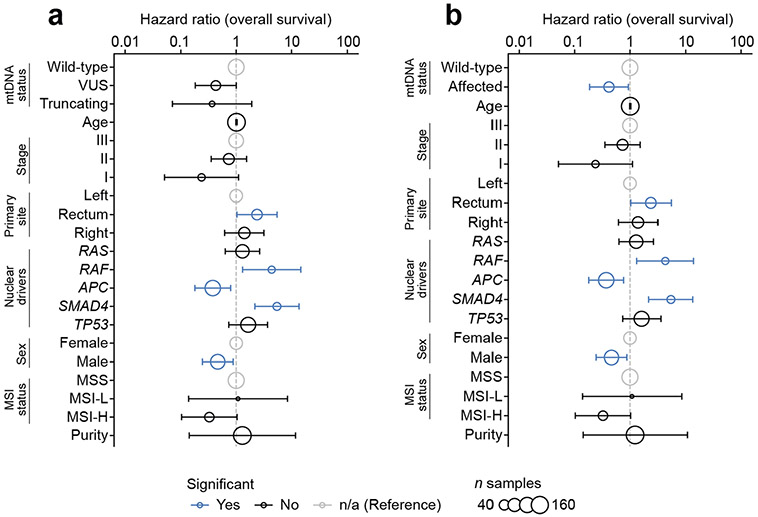
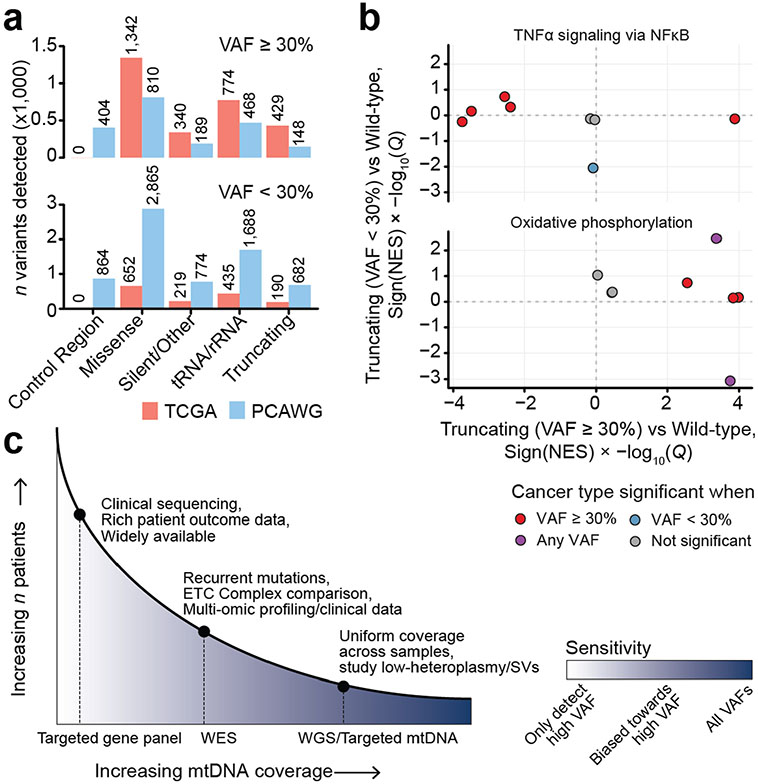
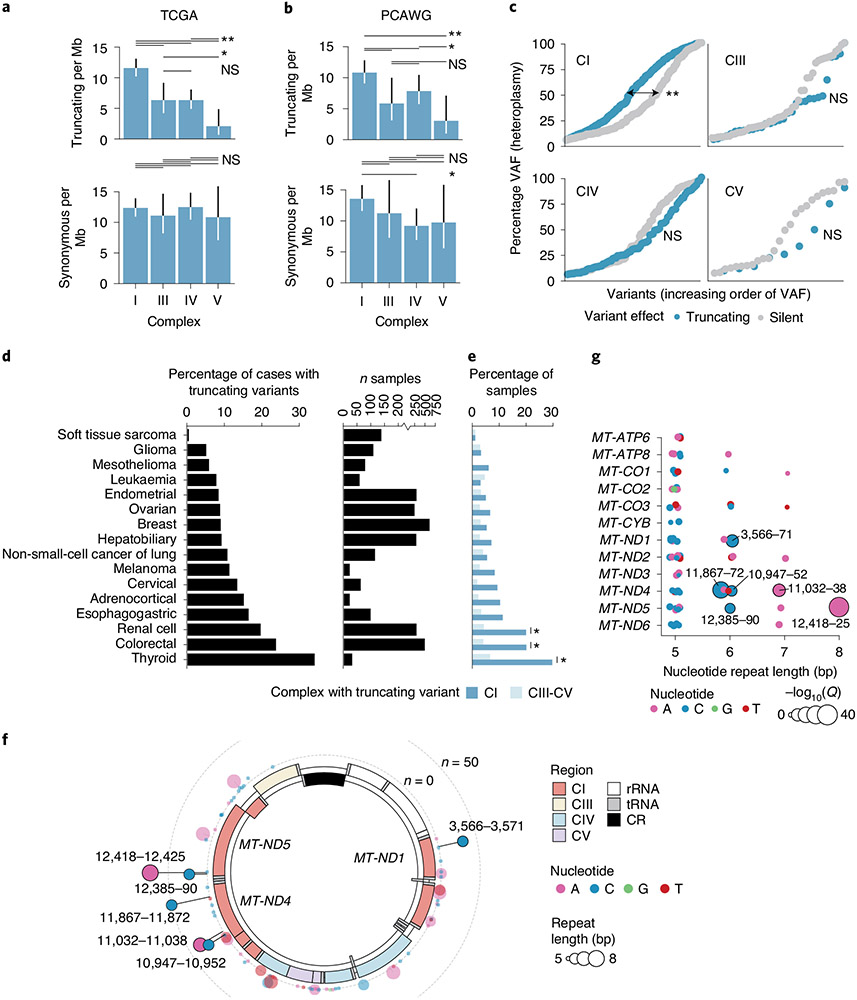
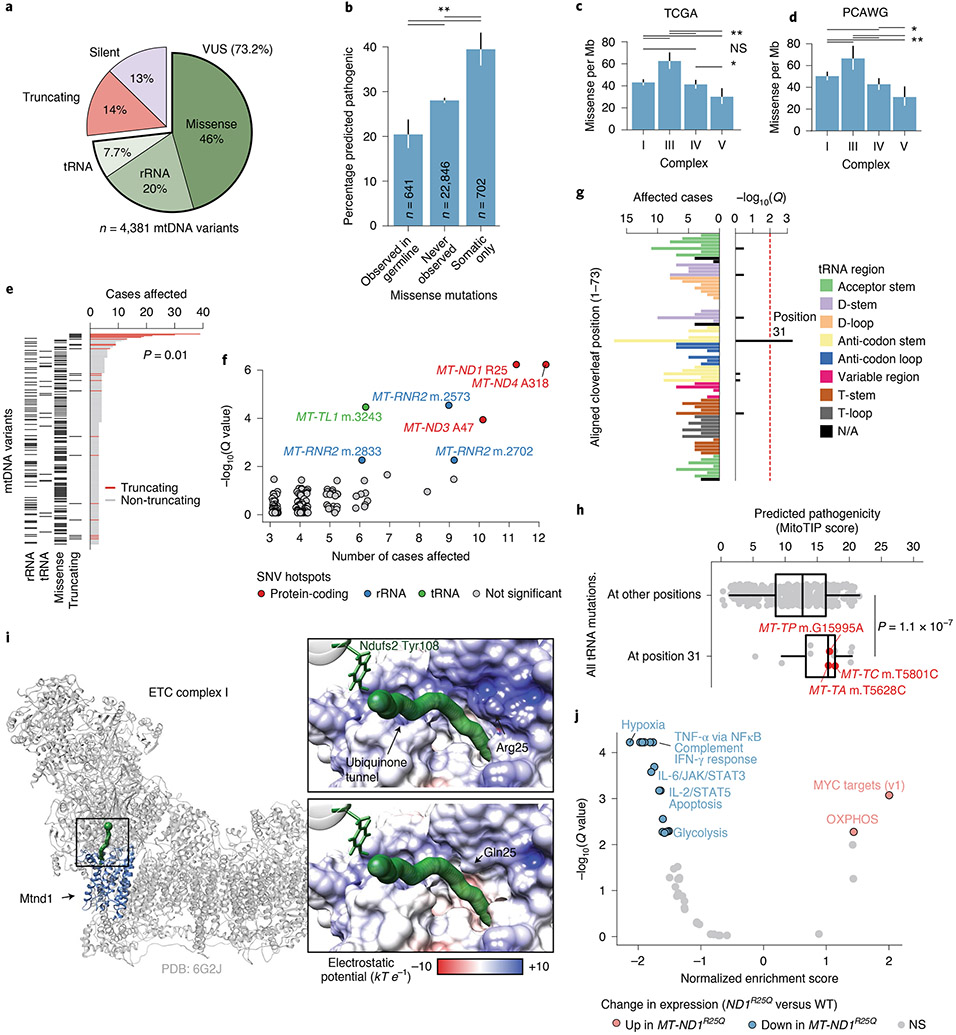
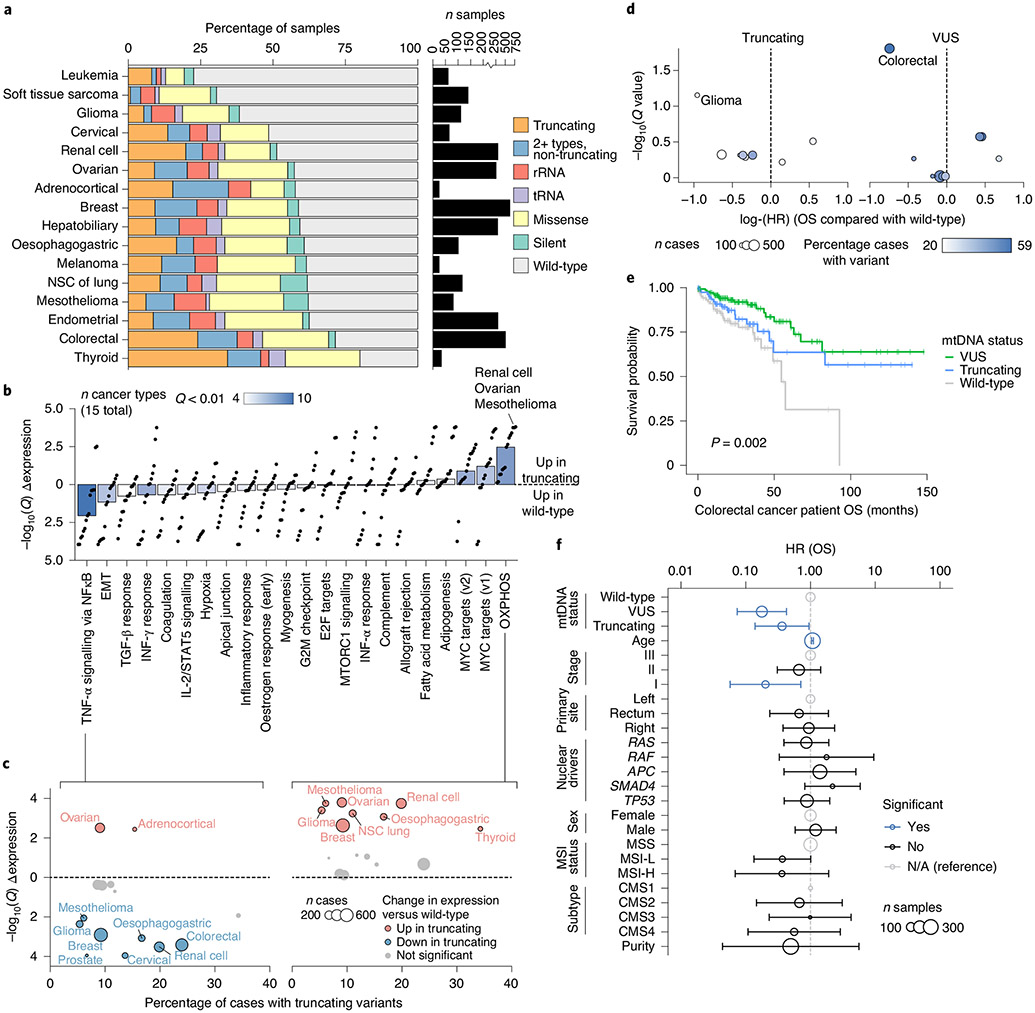
Mitochondrial DNA (mtDNA) encodes protein subunits and translational machinery required for oxidative phosphorylation (OXPHOS). Using repurposed whole-exome sequencing data, in the present study we demonstrate that pathogenic mtDNA mutations arise in tumours at a rate comparable to those in the most common cancer driver genes. We identify OXPHOS complexes as critical determinants shaping somatic mtDNA mutation patterns across tumour lineages. Loss-of-function mutations accumulate at an elevated rate specifically in complex I and often arise at specific homopolymeric hotspots. In contrast, complex V is depleted of all non-synonymous mutations, suggesting that impairment of ATP synthesis and mitochondrial membrane potential dissipation are under negative selection. Common truncating mutations and rarer missense alleles are both associated with a pan-lineage transcriptional programme, even in cancer types where mtDNA mutations are comparatively rare. Pathogenic mutations of mtDNA are associated with substantial increases in overall survival of colorectal cancer patients, demonstrating a clear functional relationship between genotype and phenotype. The mitochondrial genome is therefore frequently and functionally disrupted across many cancers, with major implications for patient stratification, prognosis and therapeutic development.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
