

ATSAS 3.0: expanded functionality and new tools for small-angle scattering data analysis
- PMID: 33833657
- PMCID: PMC7941305
- DOI: 10.1107/S1600576720013412
ATSAS 3.0: expanded functionality and new tools for small-angle scattering data analysis
Abstract
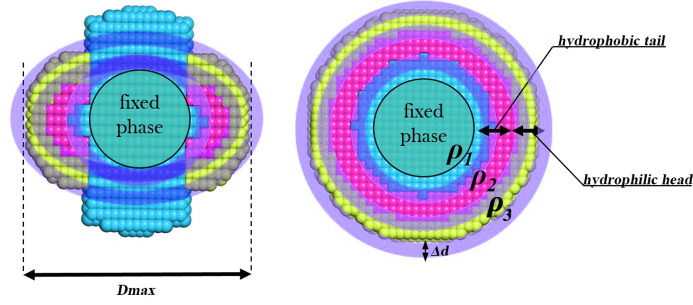
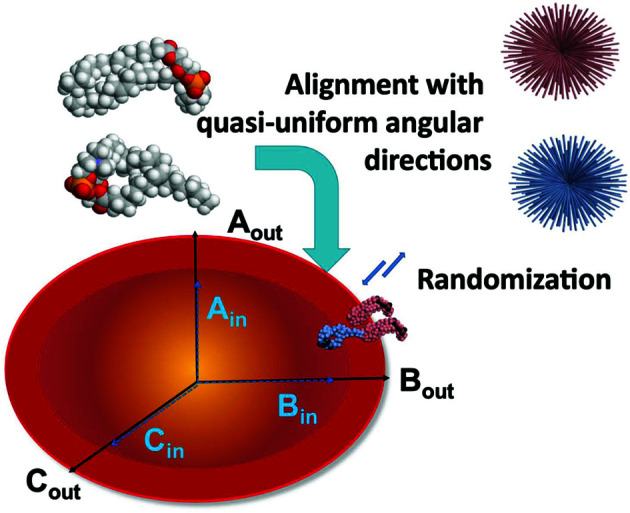
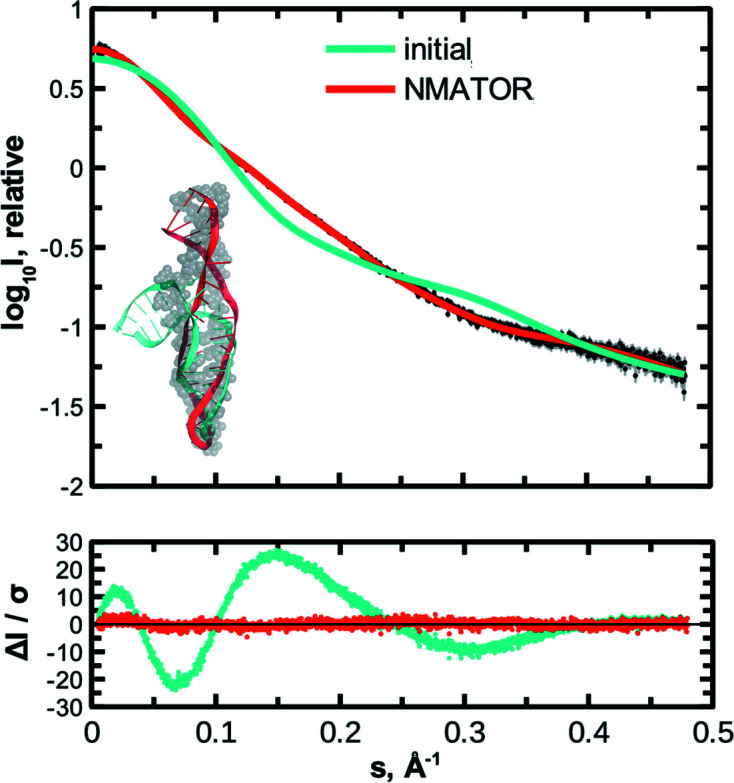
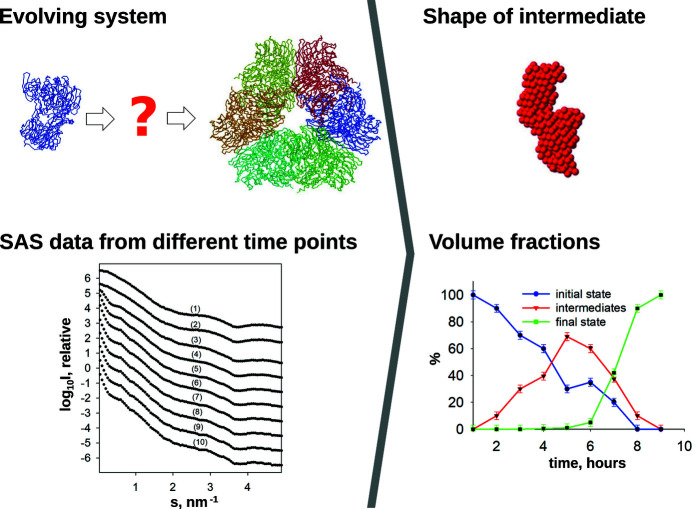
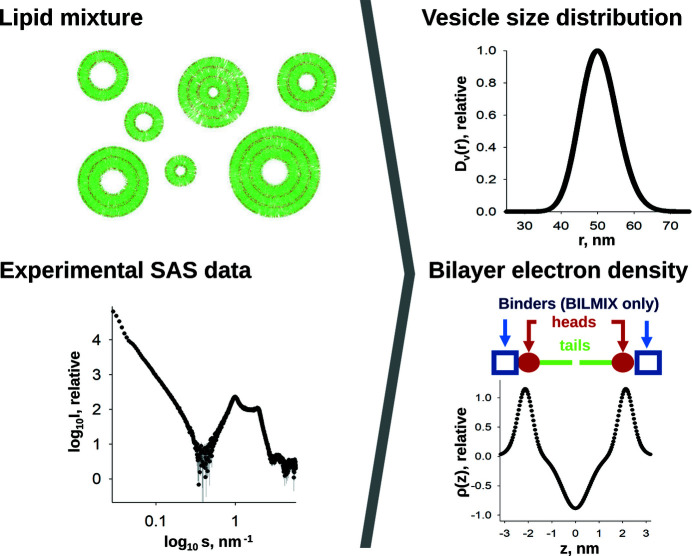
The ATSAS software suite encompasses a number of programs for the processing, visualization, analysis and modelling of small-angle scattering data, with a focus on the data measured from biological macromolecules. Here, new developments in the ATSAS 3.0 package are described. They include IMSIM, for simulating isotropic 2D scattering patterns; IMOP, to perform operations on 2D images and masks; DATRESAMPLE, a method for variance estimation of structural invariants through parametric resampling; DATFT, which computes the pair distance distribution function by a direct Fourier transform of the scattering data; PDDFFIT, to compute the scattering data from a pair distance distribution function, allowing comparison with the experimental data; a new module in DATMW for Bayesian consensus-based concentration-independent molecular weight estimation; DATMIF, an ab initio shape analysis method that optimizes the search model directly against the scattering data; DAMEMB, an application to set up the initial search volume for multiphase modelling of membrane proteins; ELLLIP, to perform quasi-atomistic modelling of liposomes with elliptical shapes; NMATOR, which models conformational changes in nucleic acid structures through normal mode analysis in torsion angle space; DAMMIX, which reconstructs the shape of an unknown intermediate in an evolving system; and LIPMIX and BILMIX, for modelling multilamellar and asymmetric lipid vesicles, respectively. In addition, technical updates were deployed to facilitate maintainability of the package, which include porting the PRIMUS graphical interface to Qt5, updating SASpy - a PyMOL plugin to run a subset of ATSAS tools - to be both Python 2 and 3 compatible, and adding utilities to facilitate mmCIF compatibility in future ATSAS releases. All these features are implemented in ATSAS 3.0, freely available for academic users at https://www.embl-hamburg.de/biosaxs/software.html.
Keywords: ATSAS; biological macromolecules; data analysis; small-angle scattering; structural modelling.
© Karen Manalastas-Cantos et al. 2021.
Figures
References
-
- Adams, P. D., Afonine, P. V., Baskaran, K., Berman, H. M., Berrisford, J., Bricogne, G., Brown, D. G., Burley, S. K., Chen, M., Feng, Z., Flensburg, C., Gutmanas, A., Hoch, J. C., Ikegawa, Y., Kengaku, Y., Krissinel, E., Kurisu, G., Liang, Y., Liebschner, D., Mak, L., Markley, J. L., Moriarty, N. W., Murshudov, G. N., Noble, M., Peisach, E., Persikova, I., Poon, B. K., Sobolev, O. V., Ulrich, E. L., Velankar, S., Vonrhein, C., Westbrook, J., Wojdyr, M., Yokochi, M. & Young, J. Y. (2019). Acta Cryst. D75, 451–454. - PMC - PubMed
-
- Anderson, T. W. & Darling, D. A. (1954). J. Am. Stat. Assoc. 49, 765–769.
-
- Anscombe, F. J. (1948). Biometrika, 35, 246–254.
LinkOut - more resources
Full Text Sources
Other Literature Sources