

Case Report: Novel SAVI-Causing Variants in STING1 Expand the Clinical Disease Spectrum and Suggest a Refined Model of STING Activation
- PMID: 33833757
- PMCID: PMC8023226
- DOI: 10.3389/fimmu.2021.636225
Case Report: Novel SAVI-Causing Variants in STING1 Expand the Clinical Disease Spectrum and Suggest a Refined Model of STING Activation
Abstract
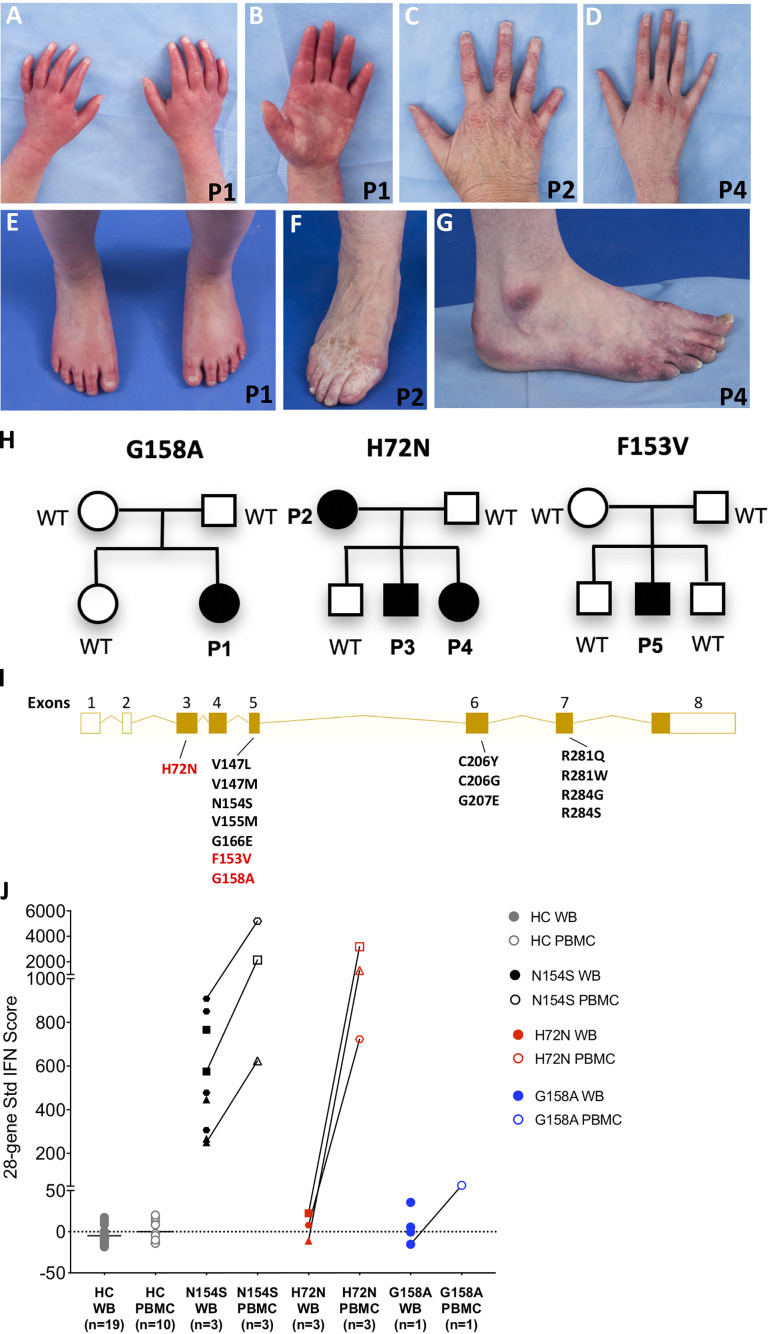
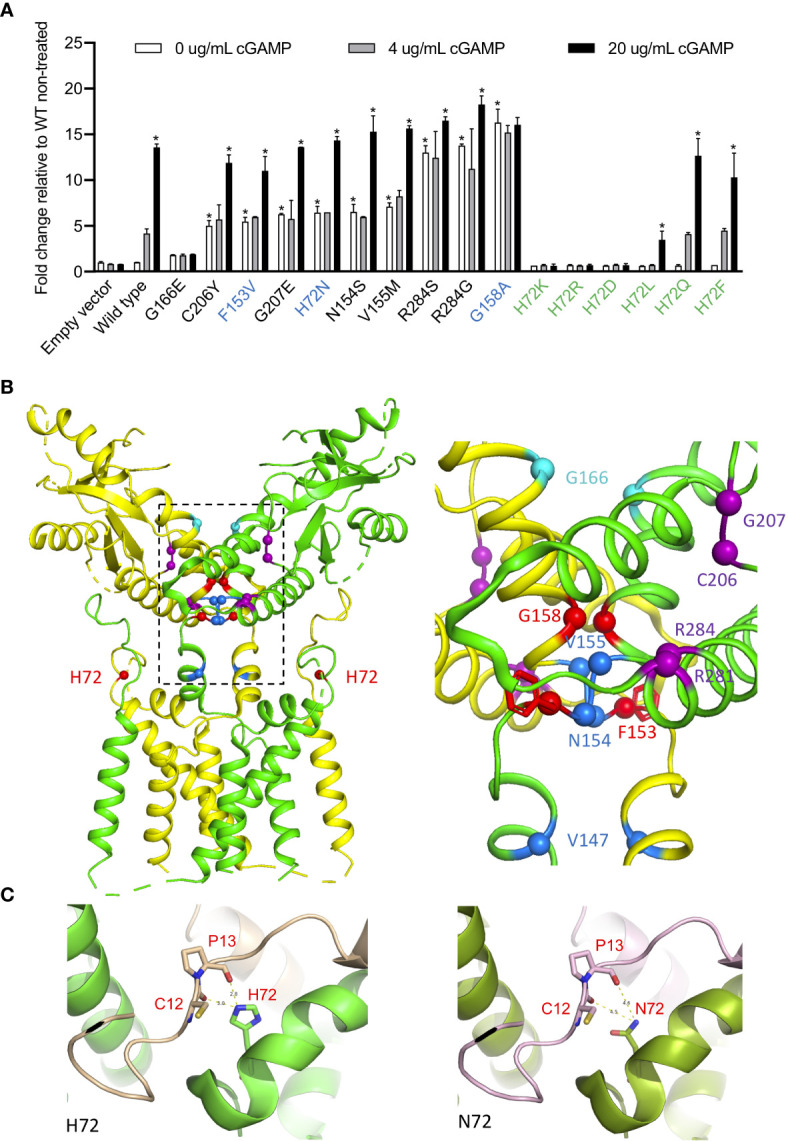
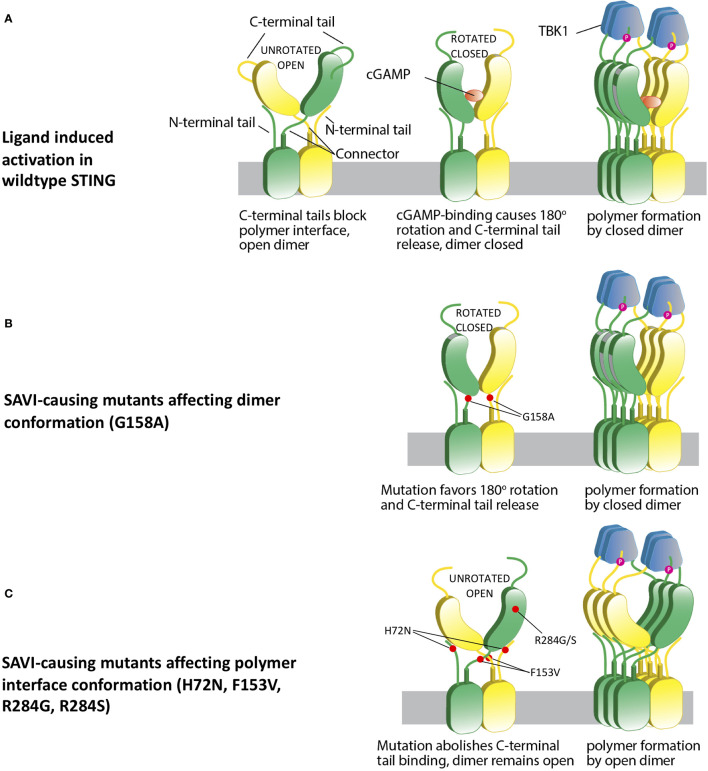
Gain-of-function mutations in STING1 cause the monogenic interferonopathy, SAVI, which presents with early-onset systemic inflammation, cold-induced vasculopathy and/or interstitial lung disease. We identified 5 patients (3 kindreds) with predominantly peripheral vascular disease who harbor 3 novel STING1 variants, p.H72N, p.F153V, and p.G158A. The latter two were predicted by a previous cryo-EM structure model to cause STING autoactivation. The p.H72N variant in exon 3, however, is the first SAVI-causing variant in the transmembrane linker region. Mutations of p.H72 into either charged residues or hydrophobic residues all led to dramatic loss of cGAMP response, while amino acid changes to residues with polar side chains were able to maintain the wild type status. Structural modeling of these novel mutations suggests a reconciled model of STING activation, which indicates that STING dimers can oligomerize in both open and closed states which would obliviate a high-energy 180° rotation of the ligand-binding head for STING activation, thus refining existing models of STING activation. Quantitative comparison showed that an overall lower autoactivating potential of the disease-causing mutations was associated with less severe lung disease, more severe peripheral vascular disease and the absence of a robust interferon signature in whole blood. Our findings are important in understanding genotype-phenotype correlation, designing targeted STING inhibitors and in dissecting differentially activated pathways downstream of different STING mutations.
Keywords: SAVI; STING; autoinflammatory disease; interferonopathy; pediatrics; type I interferon; whole exome sequencing.
Copyright © 2021 Lin, Torreggiani, Kahle, Rumsey, Wright, Montes-Cano, Silveira, Alehashemi, Mitchell, Aue, Ji, Jin, de Jesus and Goldbach-Mansky.
Conflict of interest statement
RG-M has received grant support from SOBI, Regeneron, Novartis and Eli Lilly. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
A Novel Biallelic STING1 Gene Variant Causing SAVI in Two Siblings.Front Immunol. 2021 Jan 8;11:599564. doi: 10.3389/fimmu.2020.599564. eCollection 2020. Front Immunol. 2021. PMID: 33488593 Free PMC article.
-
Overview of STING-Associated Vasculopathy with Onset in Infancy (SAVI) Among 21 Patients.J Allergy Clin Immunol Pract. 2021 Feb;9(2):803-818.e11. doi: 10.1016/j.jaip.2020.11.007. Epub 2020 Nov 18. J Allergy Clin Immunol Pract. 2021. PMID: 33217613
-
Severe interstitial lung disease as the first manifestation of a STING1 variant in a familial case.Immunol Res. 2025 Jul 29;73(1):113. doi: 10.1007/s12026-025-09670-1. Immunol Res. 2025. PMID: 40731159
-
Lung Inflammation in STING-Associated Vasculopathy with Onset in Infancy (SAVI).Cells. 2022 Jan 18;11(3):318. doi: 10.3390/cells11030318. Cells. 2022. PMID: 35159128 Free PMC article. Review.
-
STING-Mediated Lung Inflammation and Beyond.J Clin Immunol. 2021 Apr;41(3):501-514. doi: 10.1007/s10875-021-00974-z. Epub 2021 Feb 2. J Clin Immunol. 2021. PMID: 33532887 Review.
Cited by
-
Clathrin-associated AP-1 controls termination of STING signalling.Nature. 2022 Oct;610(7933):761-767. doi: 10.1038/s41586-022-05354-0. Epub 2022 Oct 19. Nature. 2022. PMID: 36261523 Free PMC article.
-
Emerging concepts and treatments in autoinflammatory interferonopathies and monogenic systemic lupus erythematosus.Nat Rev Rheumatol. 2025 Jan;21(1):22-45. doi: 10.1038/s41584-024-01184-8. Epub 2024 Dec 2. Nat Rev Rheumatol. 2025. PMID: 39623155 Review.
-
Human STING Is Regulated by an Autoinhibitory Mechanism for Type I Interferon Production.J Innate Immun. 2022;14(5):518-531. doi: 10.1159/000521734. Epub 2022 Feb 1. J Innate Immun. 2022. PMID: 35104824 Free PMC article.
-
Orthosteric STING inhibition elucidates molecular correction of SAVI STING.Nat Commun. 2025 Jul 1;16(1):5695. doi: 10.1038/s41467-025-60632-5. Nat Commun. 2025. PMID: 40595544 Free PMC article.
-
STING Targeting in Lung Diseases.Cells. 2022 Nov 3;11(21):3483. doi: 10.3390/cells11213483. Cells. 2022. PMID: 36359882 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials