

Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases
- PMID: 33834868
- PMCID: PMC8163660
- DOI: 10.1152/ajpheart.00917.2020
Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases
Abstract
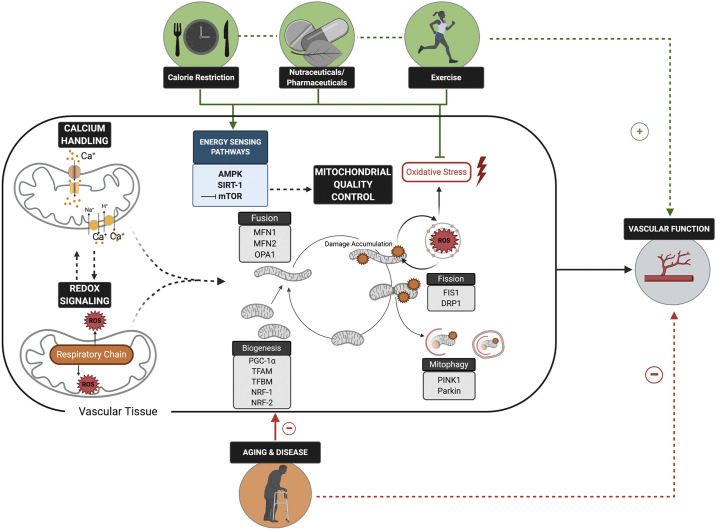
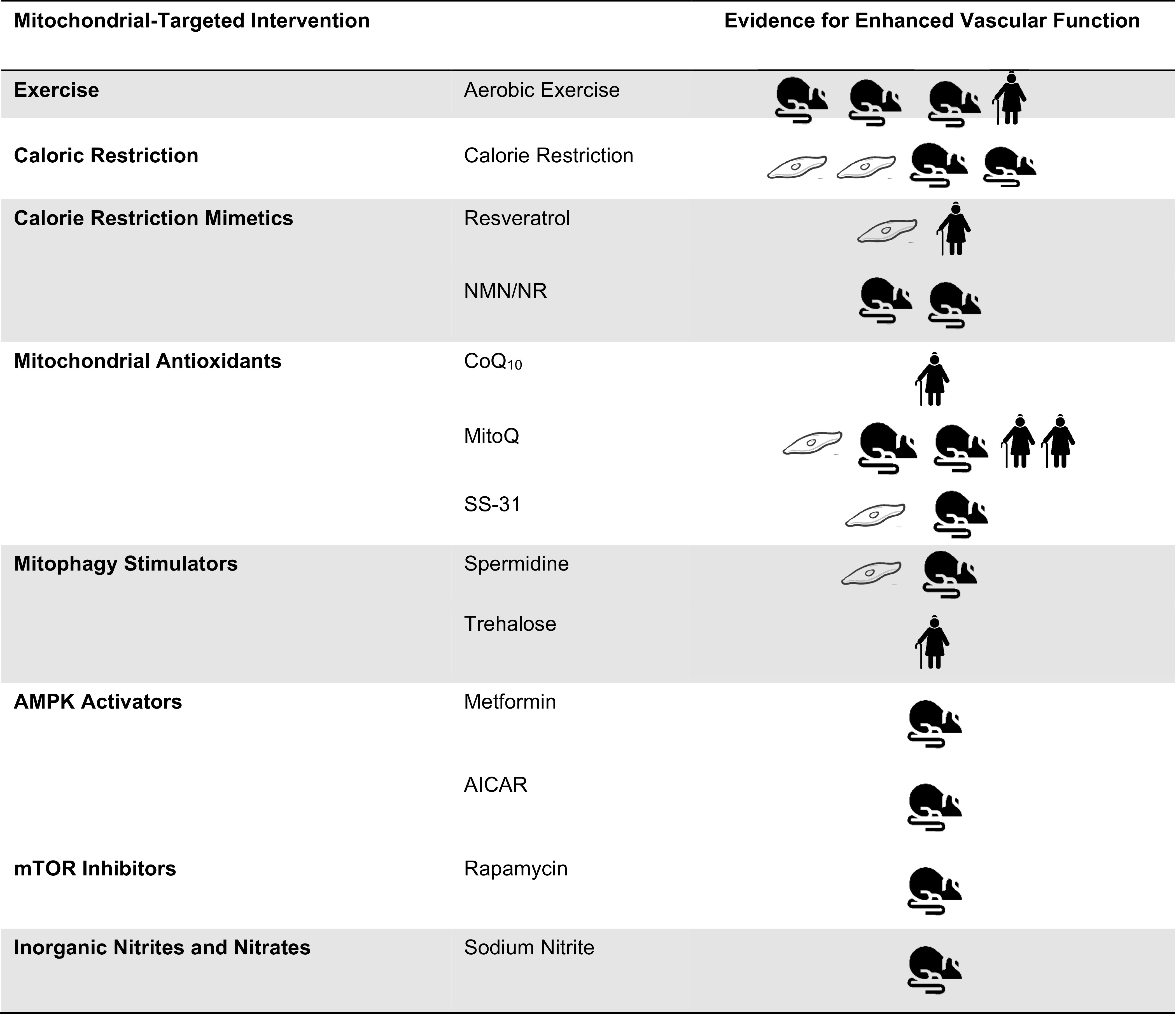
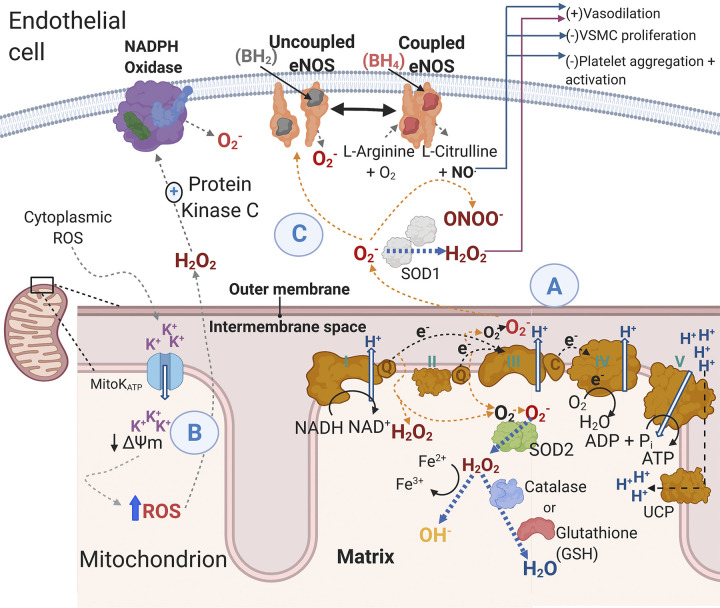
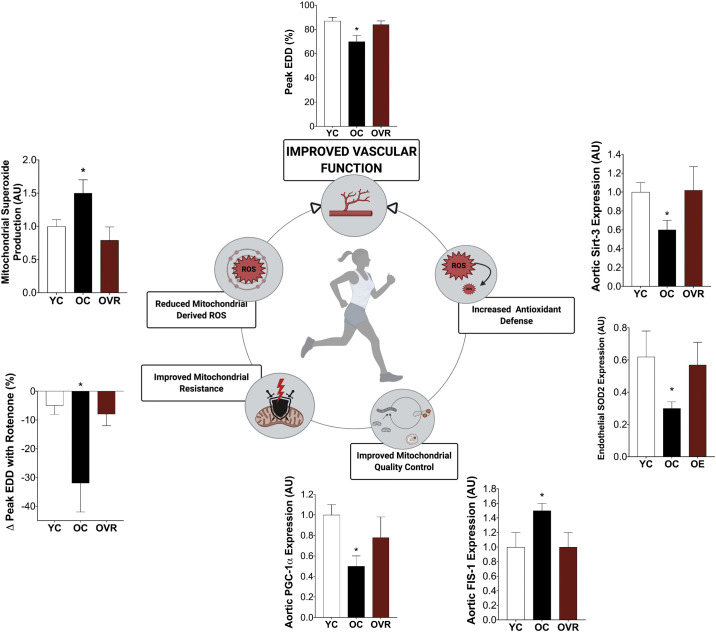
Cardiovascular disease (CVD) affects one in three adults and remains the leading cause of death in America. Advancing age is a major risk factor for CVD. Recent plateaus in CVD-related mortality rates in high-income countries after decades of decline highlight a critical need to identify novel therapeutic targets and strategies to mitigate and manage the risk of CVD development and progression. Vascular dysfunction, characterized by endothelial dysfunction and large elastic artery stiffening, is independently associated with an increased CVD risk and incidence and is therefore an attractive target for CVD prevention and management. Vascular mitochondria have emerged as an important player in maintaining vascular homeostasis. As such, age- and disease-related impairments in mitochondrial function contribute to vascular dysfunction and consequent increases in CVD risk. This review outlines the role of mitochondria in vascular function and discusses the ramifications of mitochondrial dysfunction on vascular health in the setting of age and disease. The adverse vascular consequences of increased mitochondrial-derived reactive oxygen species, impaired mitochondrial quality control, and defective mitochondrial calcium cycling are emphasized, in particular. Current evidence for both lifestyle and pharmaceutical mitochondrial-targeted strategies to improve vascular function is also presented.
Keywords: endothelium; mitochondria; vascular; vascular stiffness.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation 139: e56–e528, 2019. [Erratum in Circulation 141: e33, 2020]. - PubMed
-
- Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Piña IL, Trogdon JG, Stroke Council. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail 6: 606–619, 2013. doi: 10.1161/HHF.0b013e318291329a. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials