

Photoclick Chemistry: A Bright Idea
- PMID: 33835796
- PMCID: PMC9883840
- DOI: 10.1021/acs.chemrev.0c01212
Photoclick Chemistry: A Bright Idea
Abstract
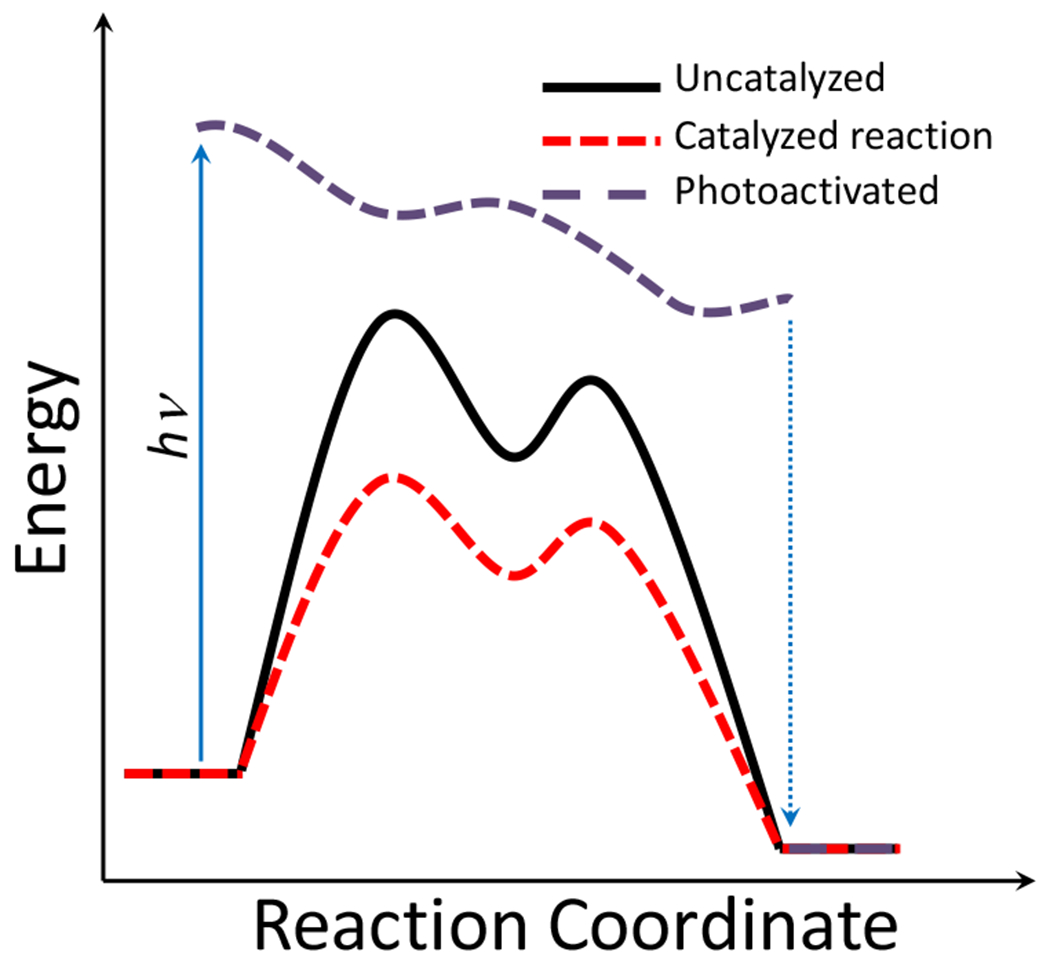
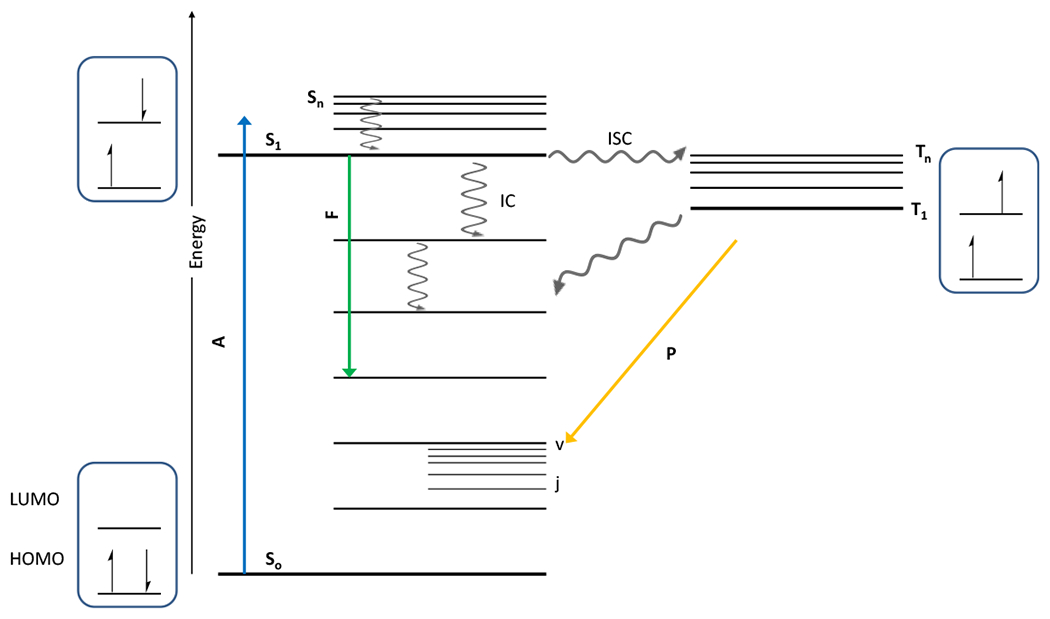
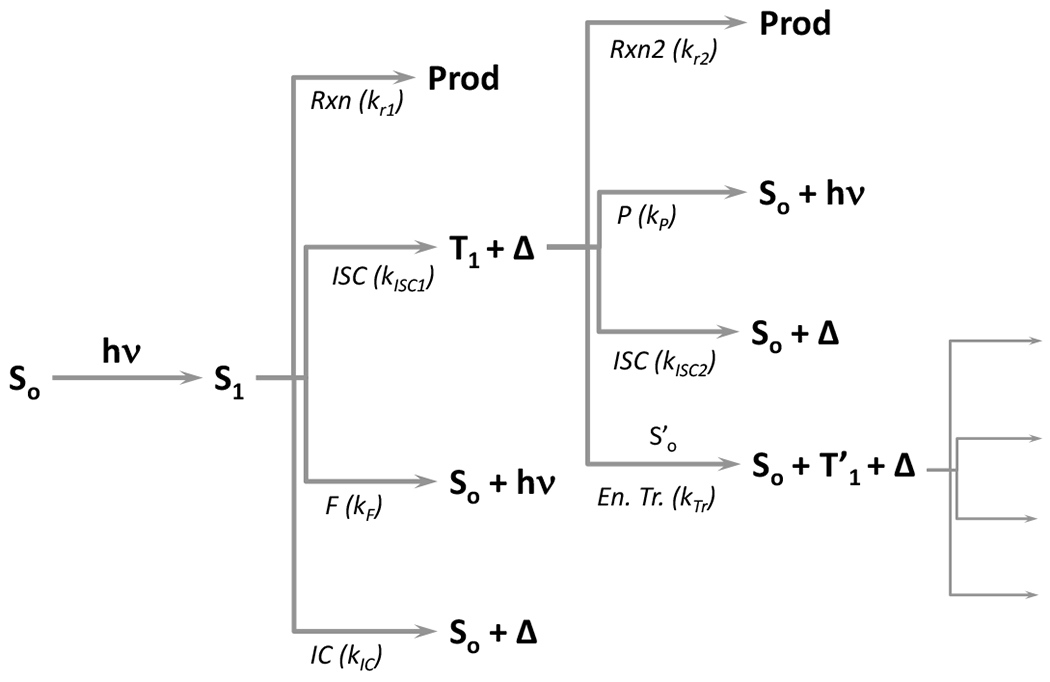
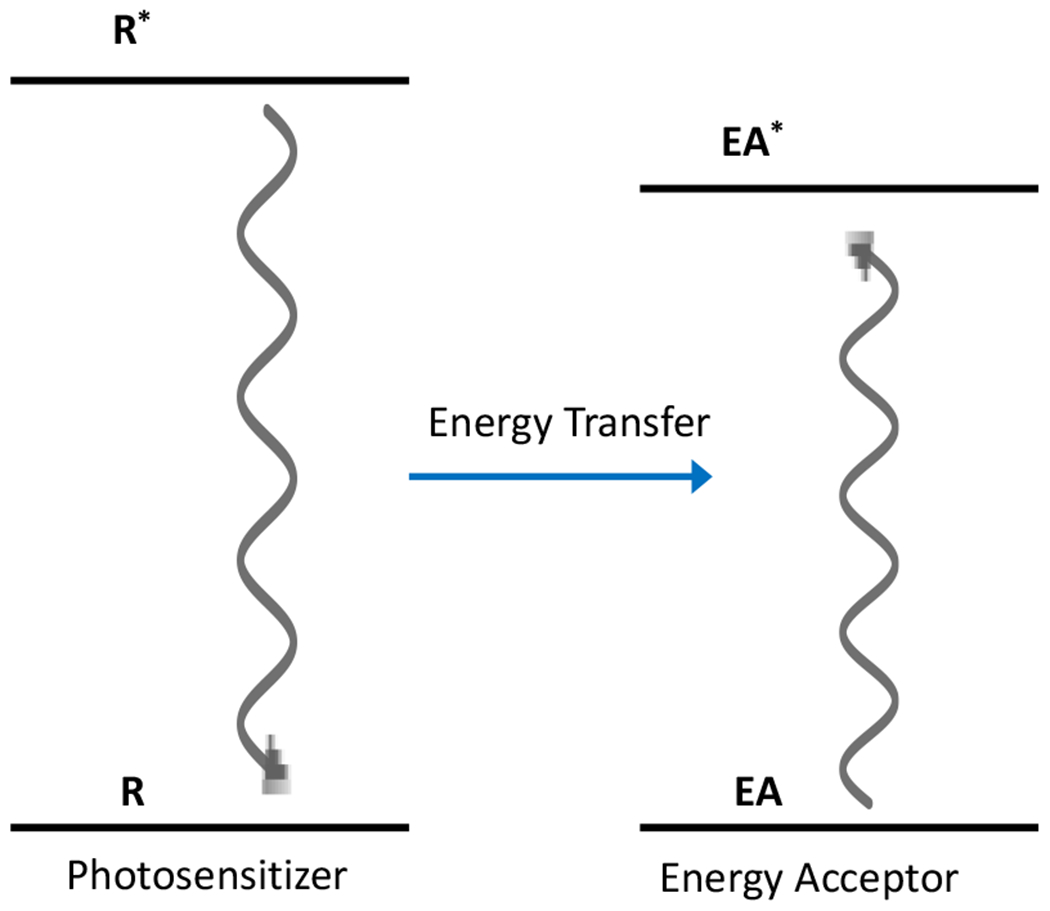
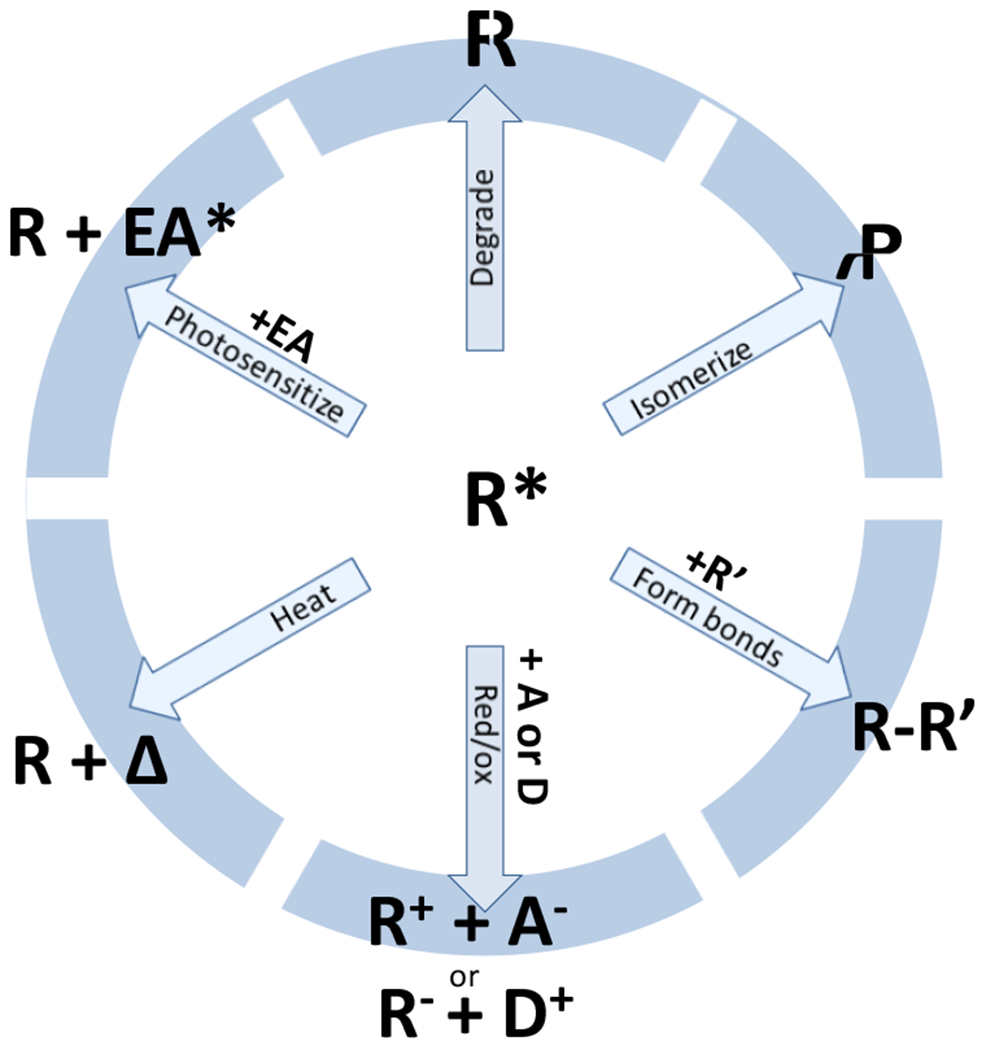
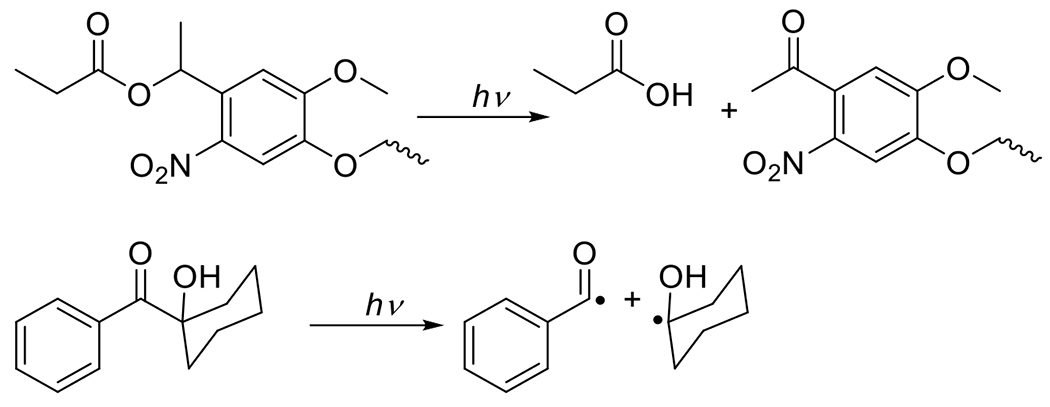
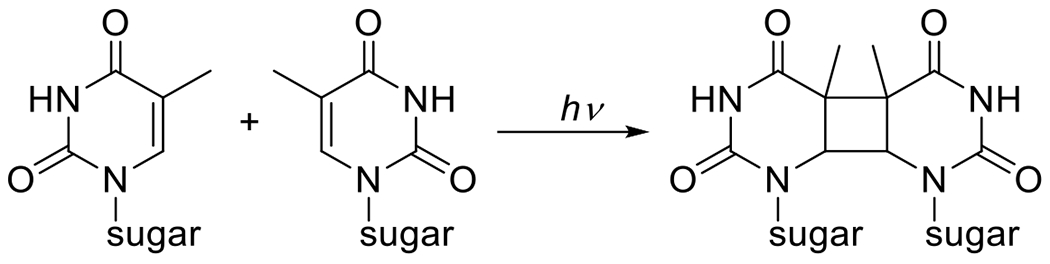
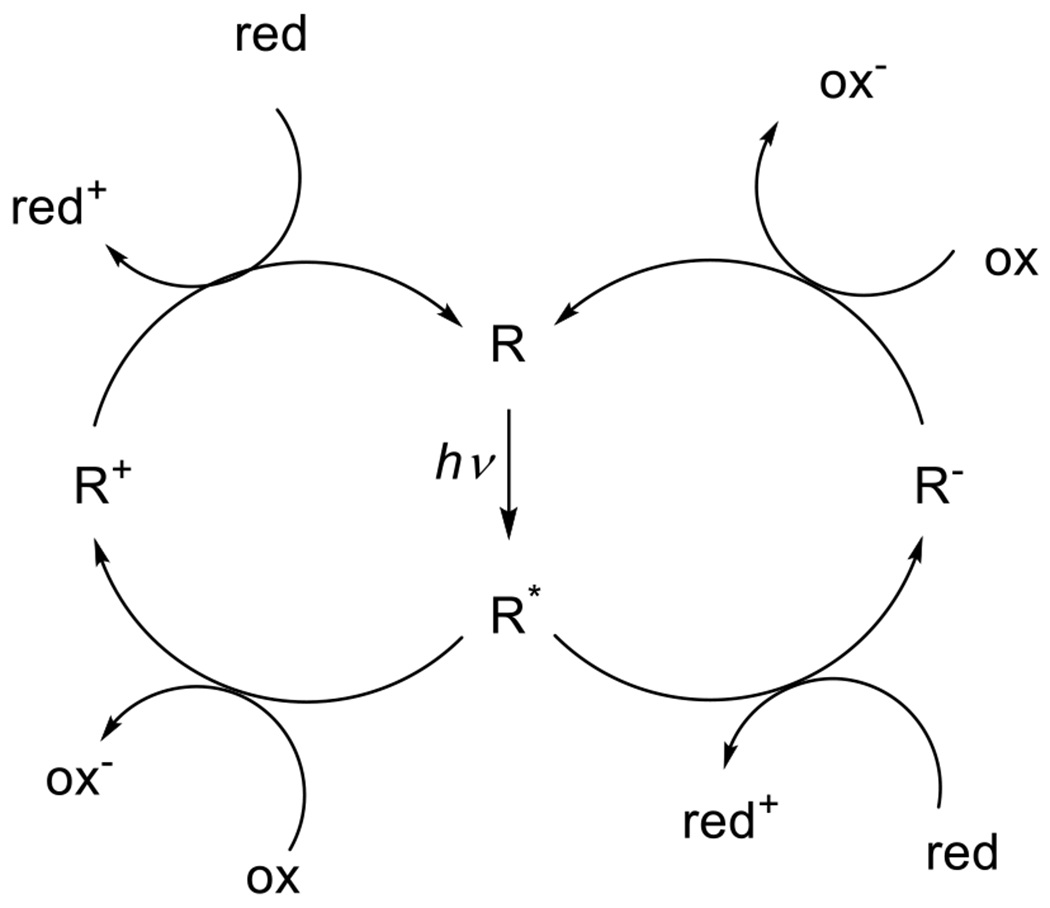
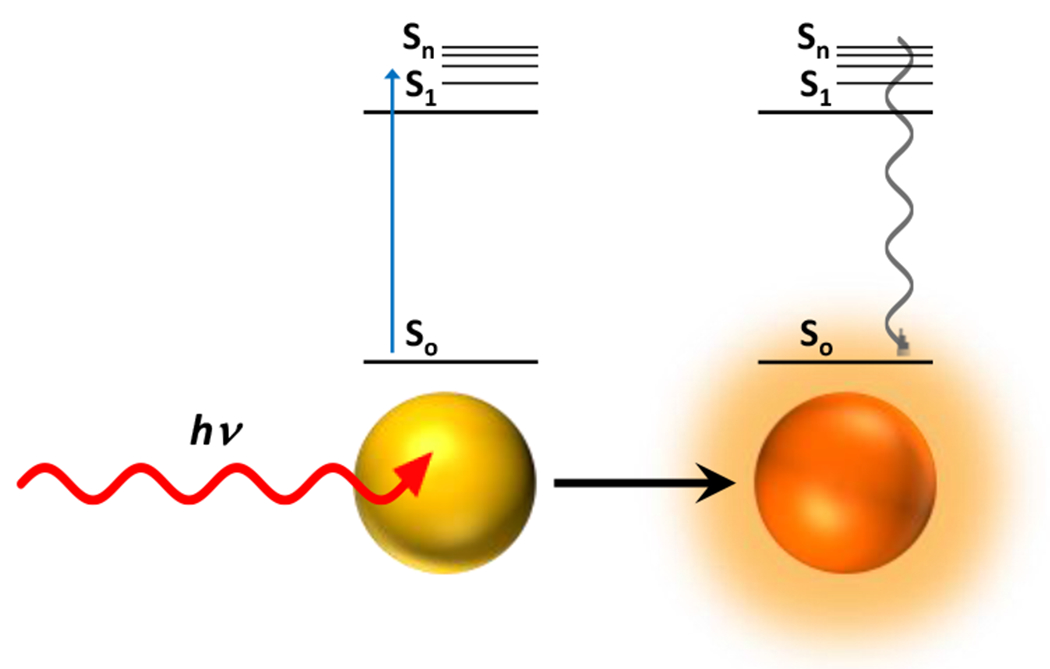
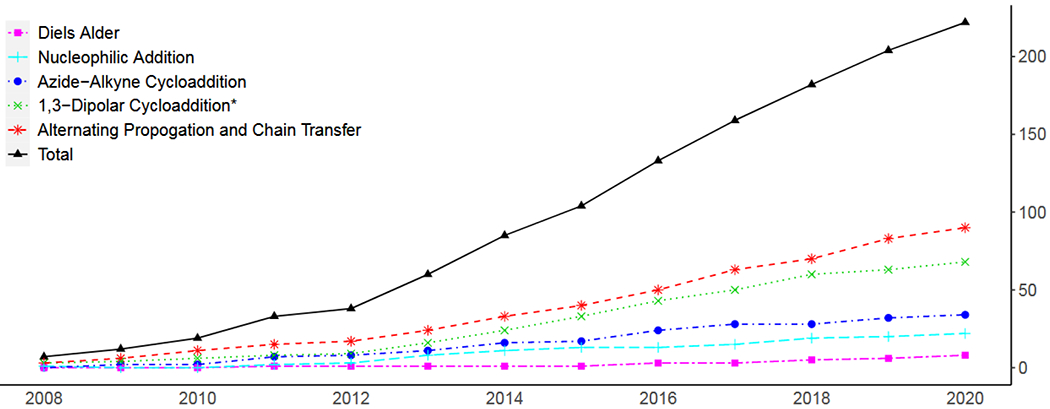
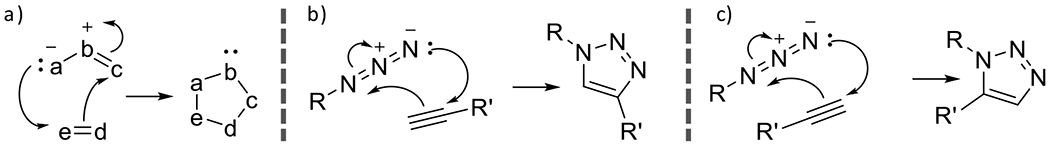
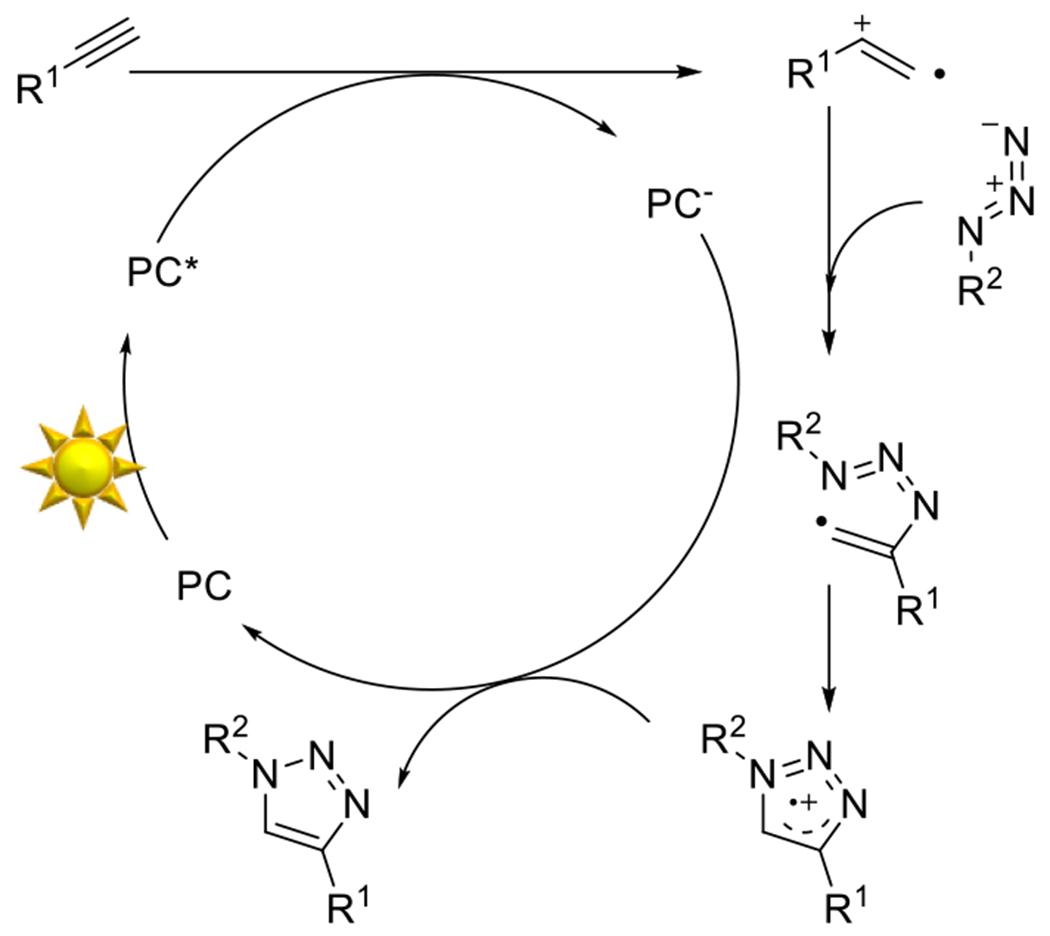
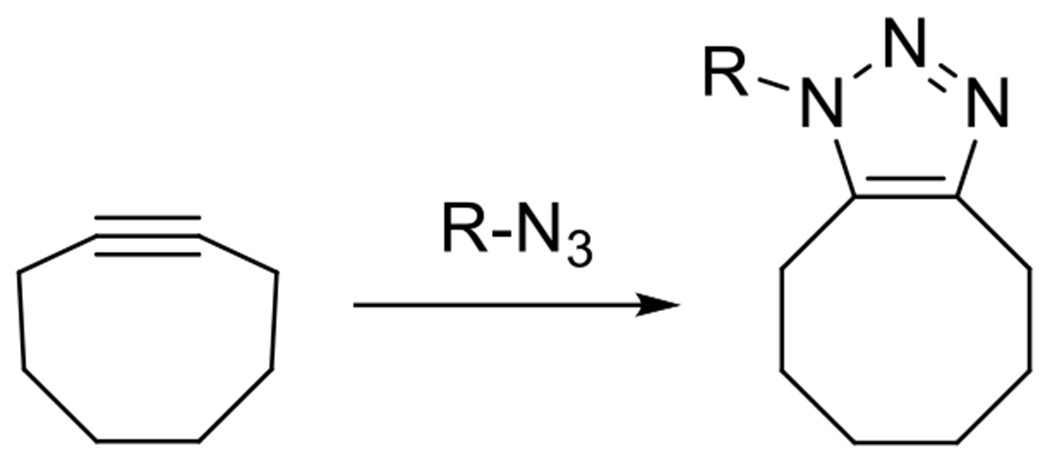
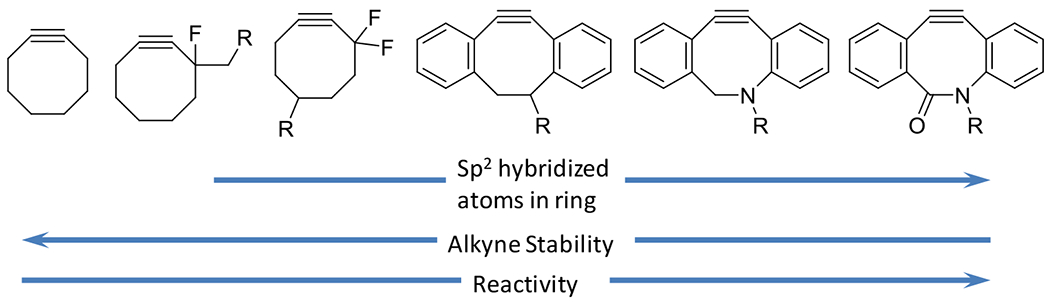
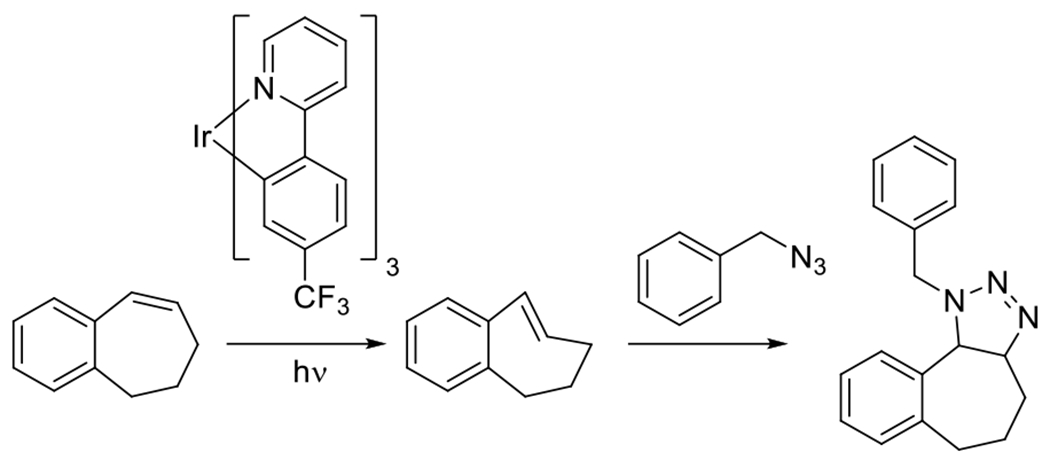
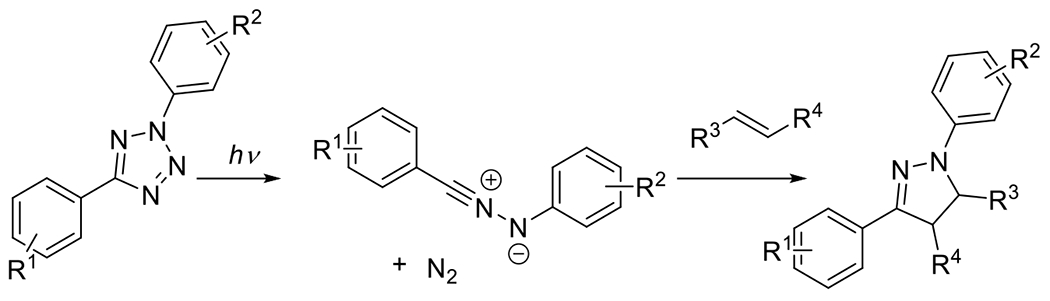
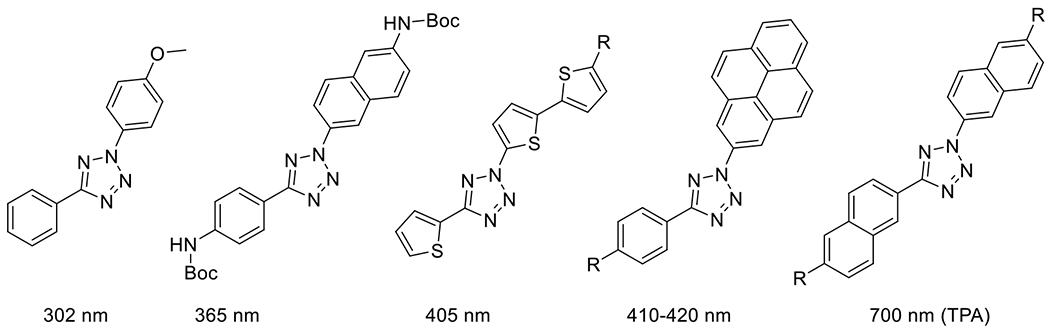
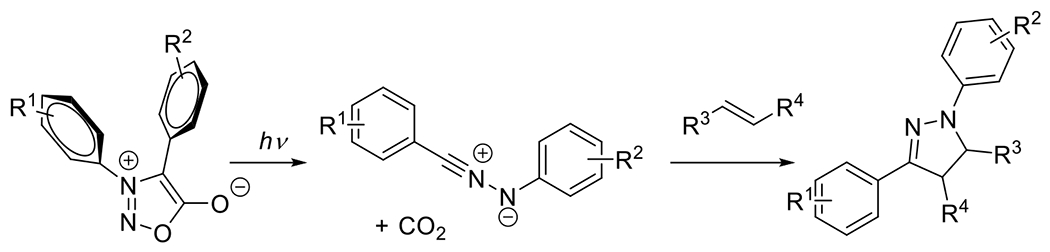
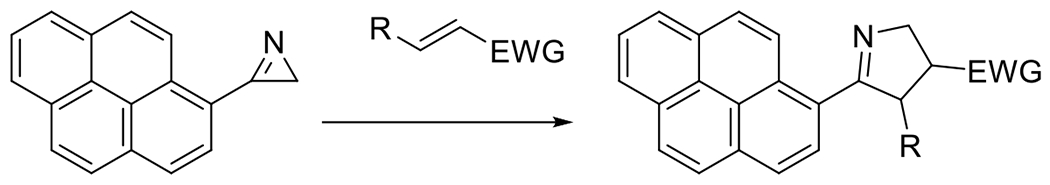
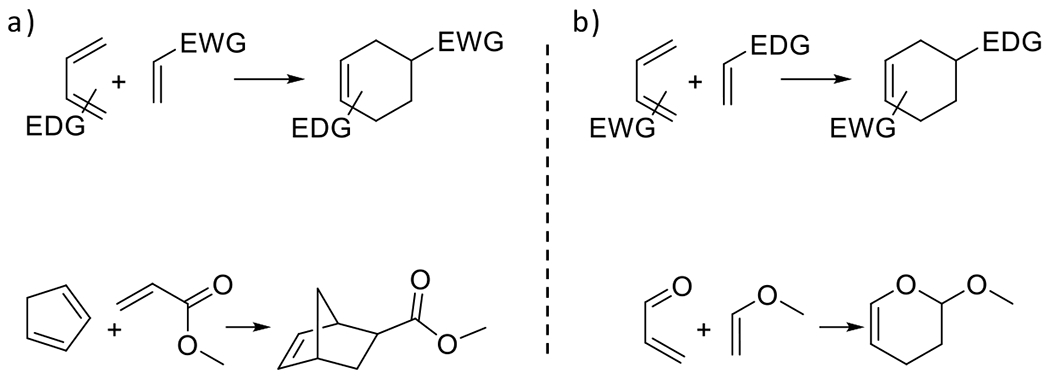
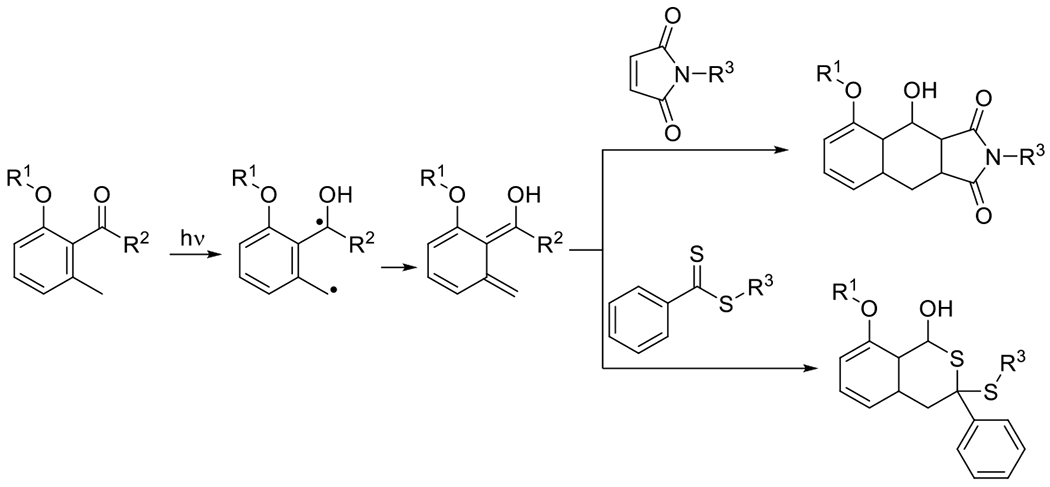
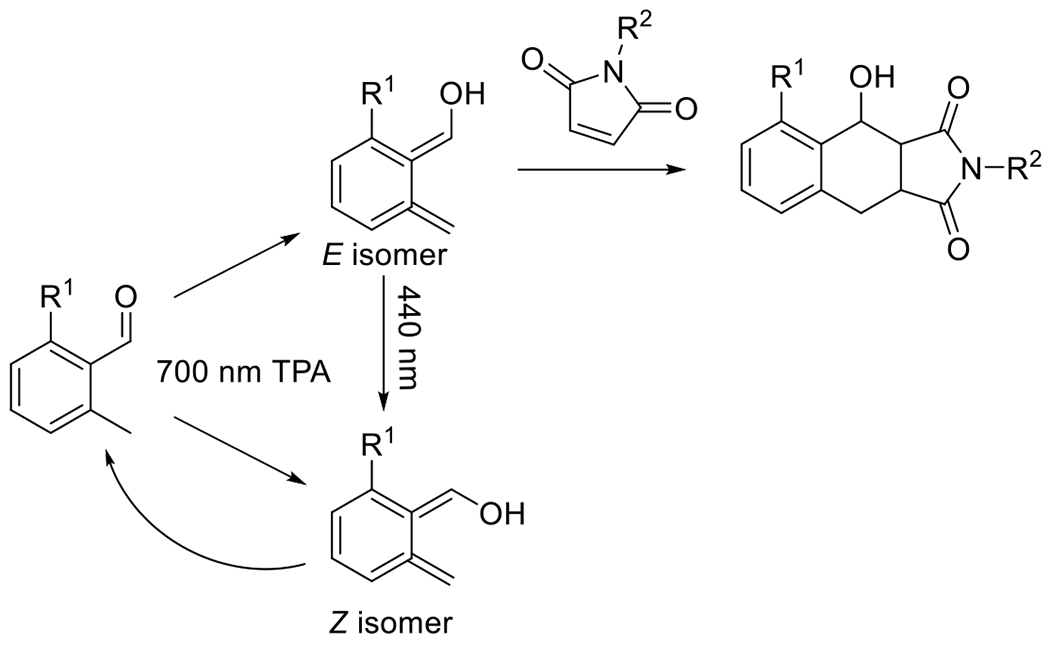
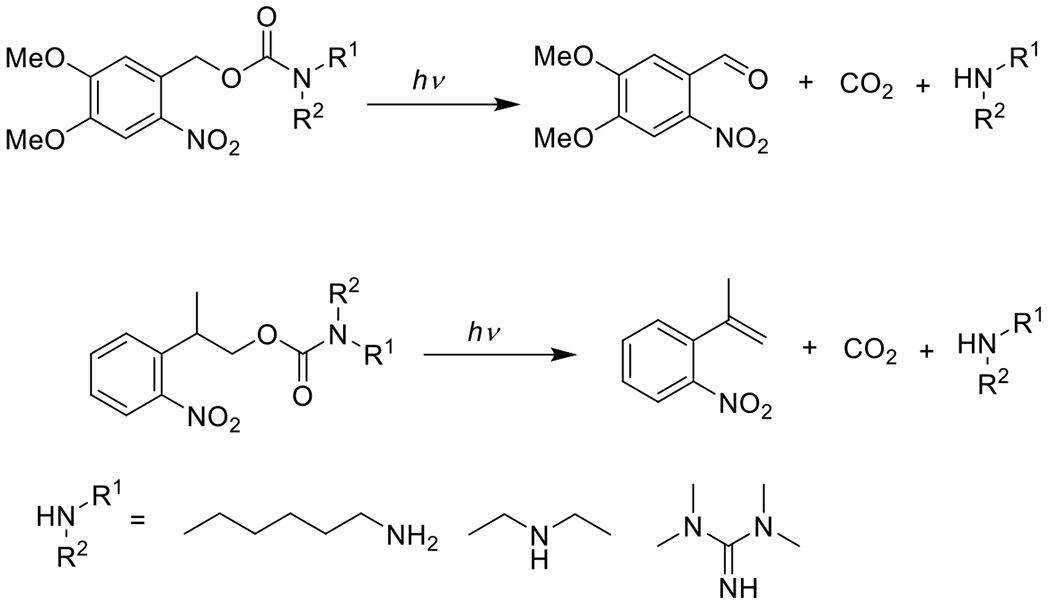
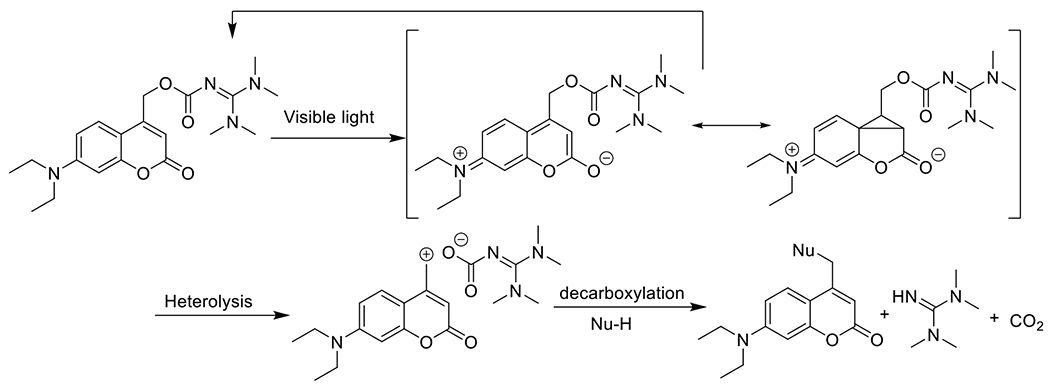
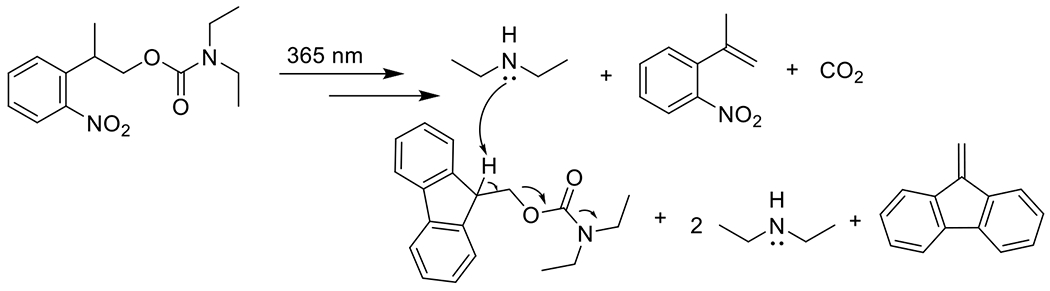
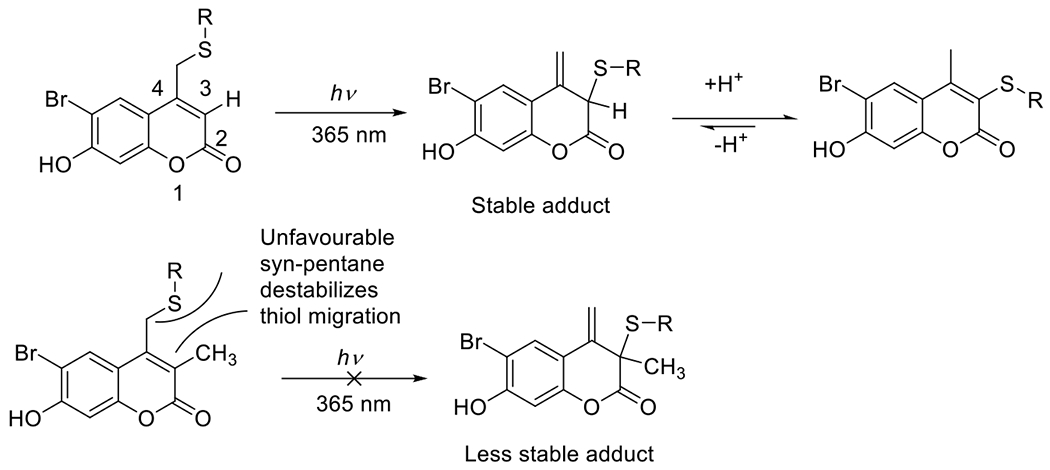
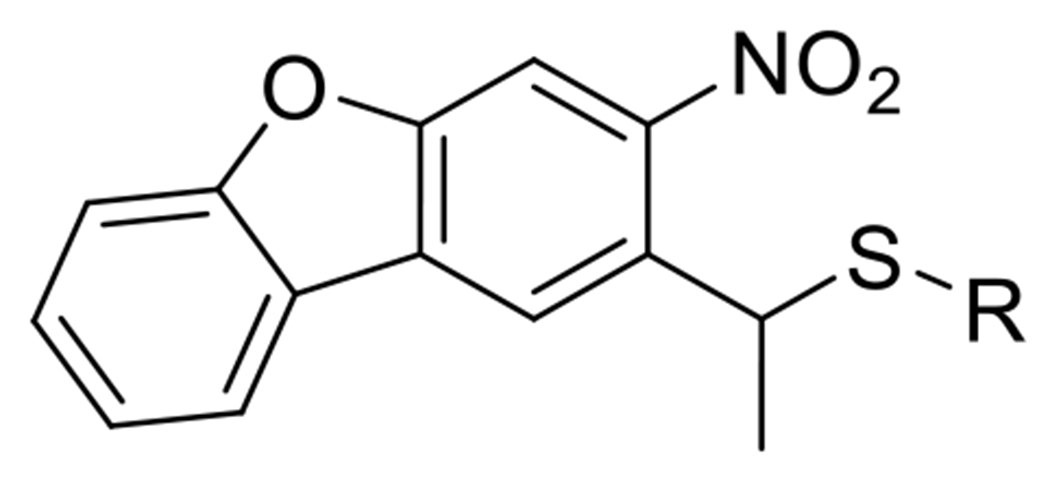
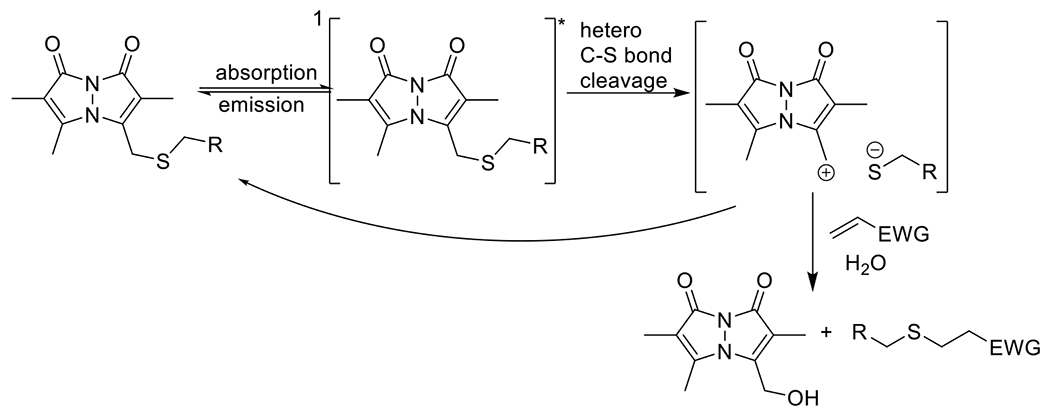
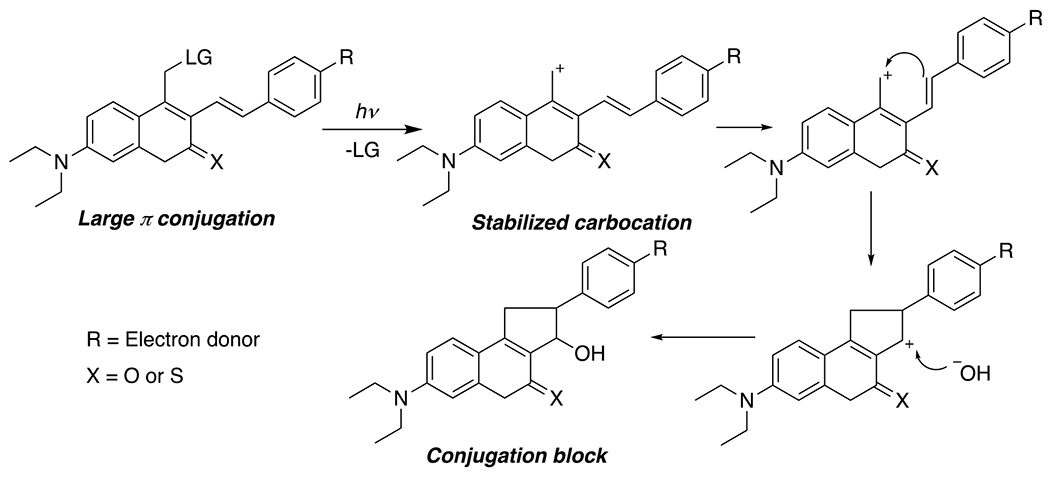
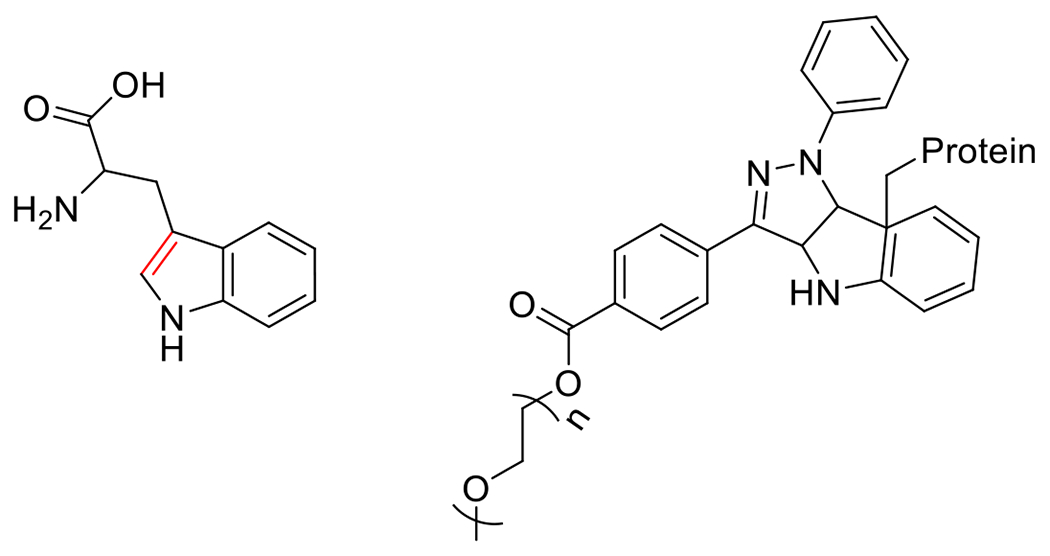
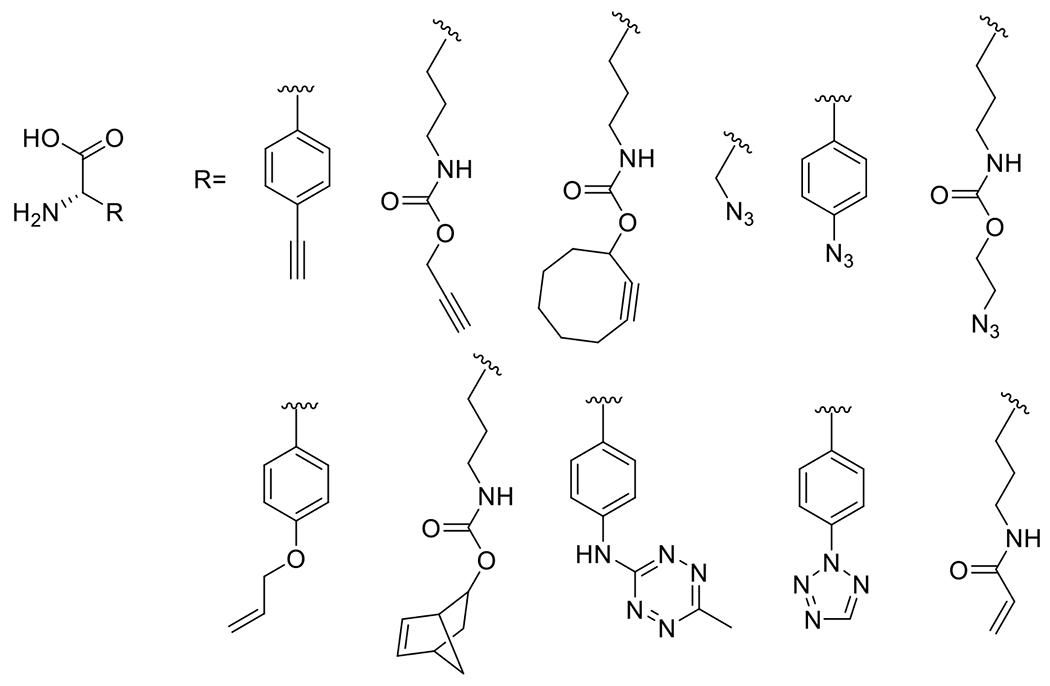
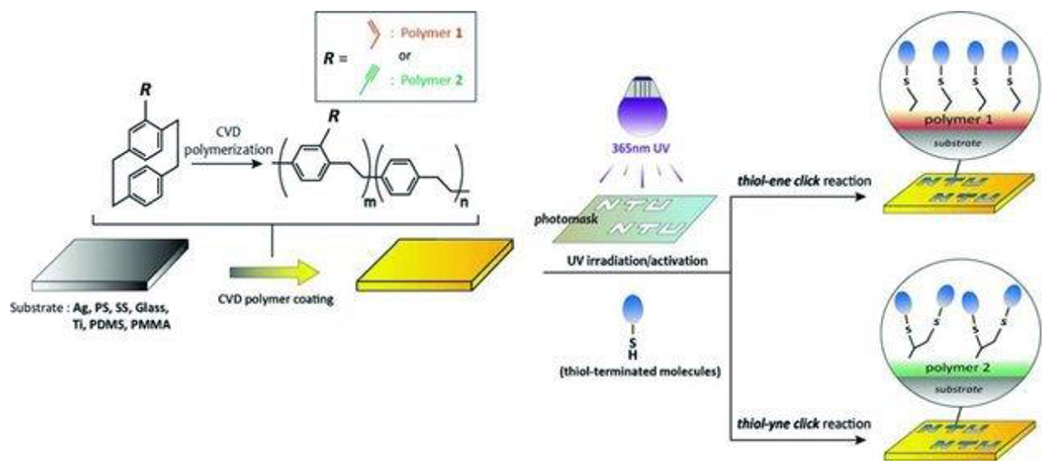
At its basic conceptualization, photoclick chemistry embodies a collection of click reactions that are performed via the application of light. The emergence of this concept has had diverse impact over a broad range of chemical and biological research due to the spatiotemporal control, high selectivity, and excellent product yields afforded by the combination of light and click chemistry. While the reactions designated as "photoclick" have many important features in common, each has its own particular combination of advantages and shortcomings. A more extensive realization of the potential of this chemistry requires a broader understanding of the physical and chemical characteristics of the specific reactions. This review discusses the features of the most frequently employed photoclick reactions reported in the literature: photomediated azide-alkyne cycloadditions, other 1,3-dipolarcycloadditions, Diels-Alder and inverse electron demand Diels-Alder additions, radical alternating addition chain transfer additions, and nucleophilic additions. Applications of these reactions in a variety of chemical syntheses, materials chemistry, and biological contexts are surveyed, with particular attention paid to the respective strengths and limitations of each reaction and how that reaction benefits from its combination with light. Finally, challenges to broader employment of these reactions are discussed, along with strategies and opportunities to mitigate such obstacles.
Conflict of interest statement
The authors declare no competing financial interest.
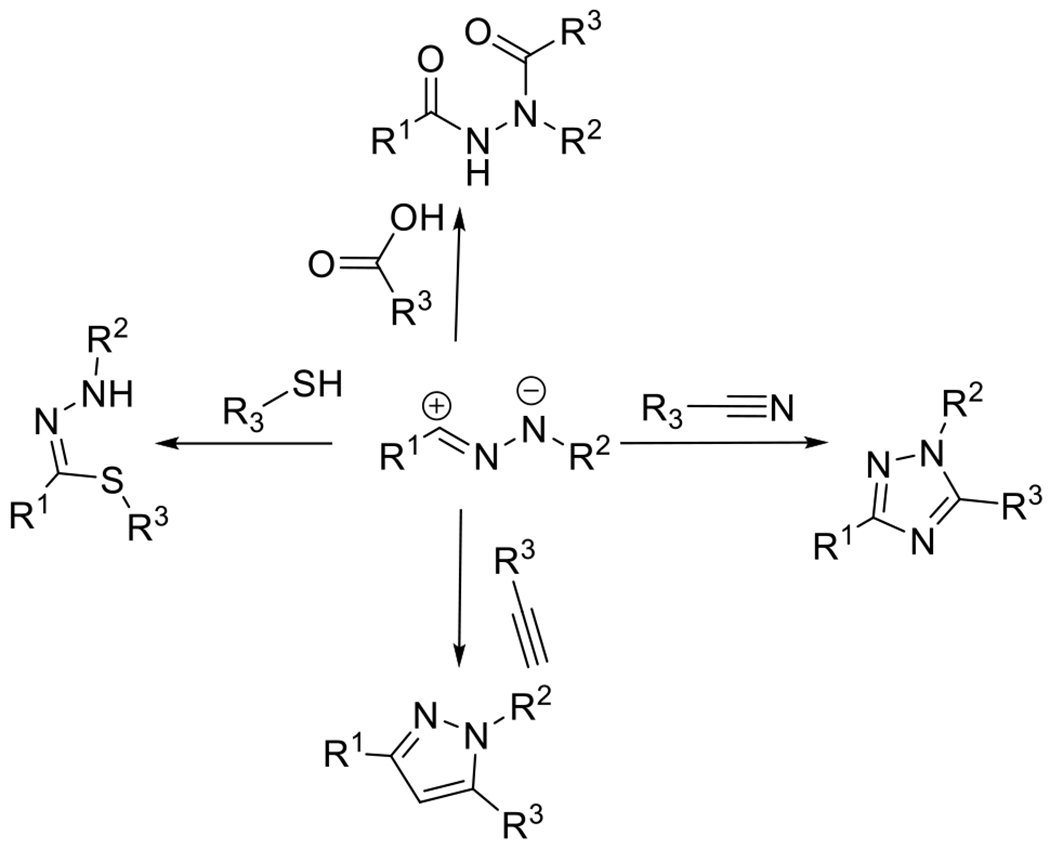
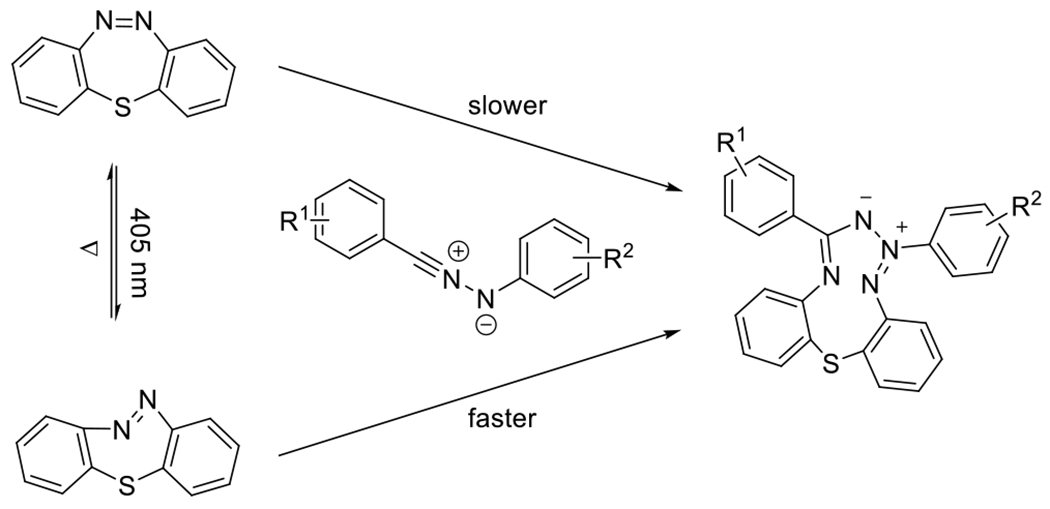
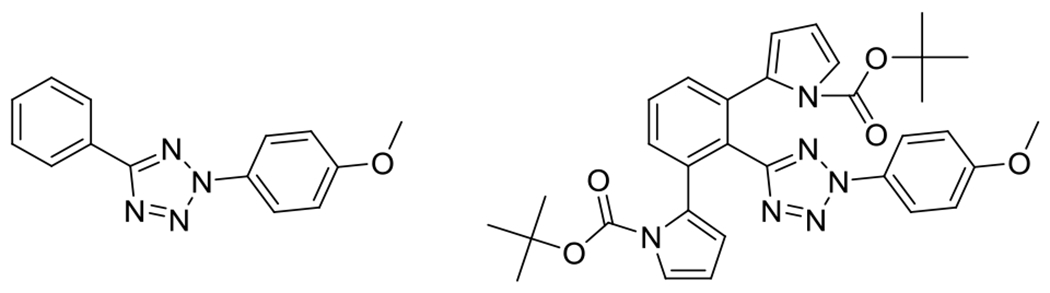
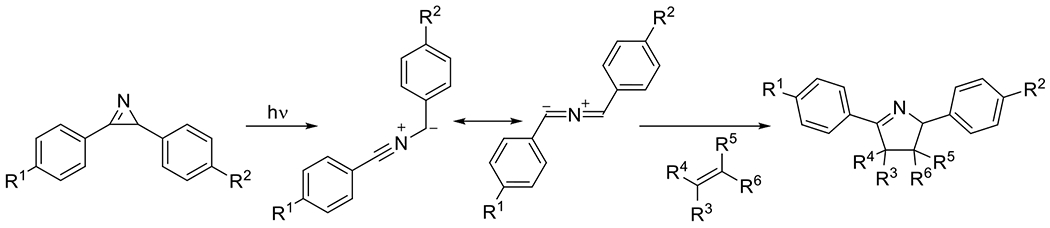
Figures



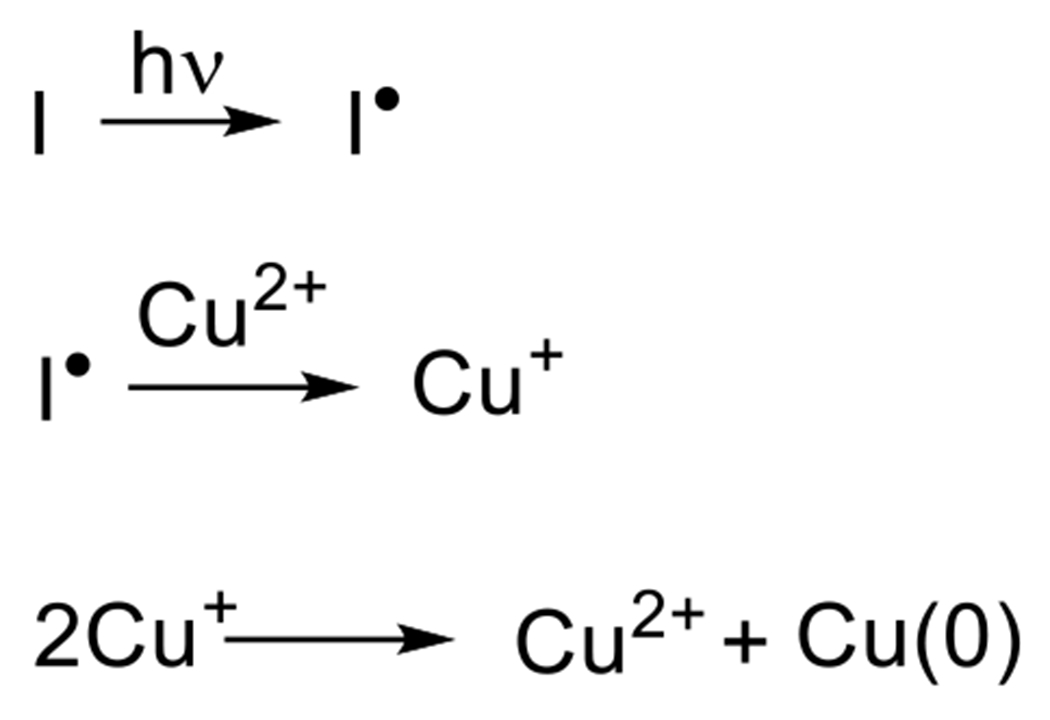



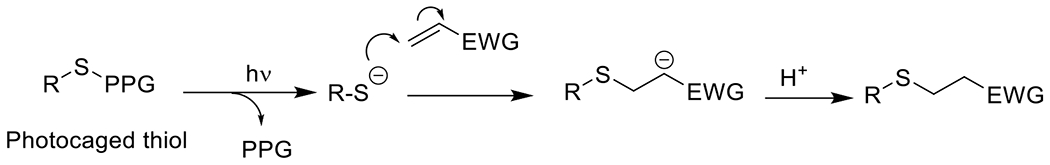
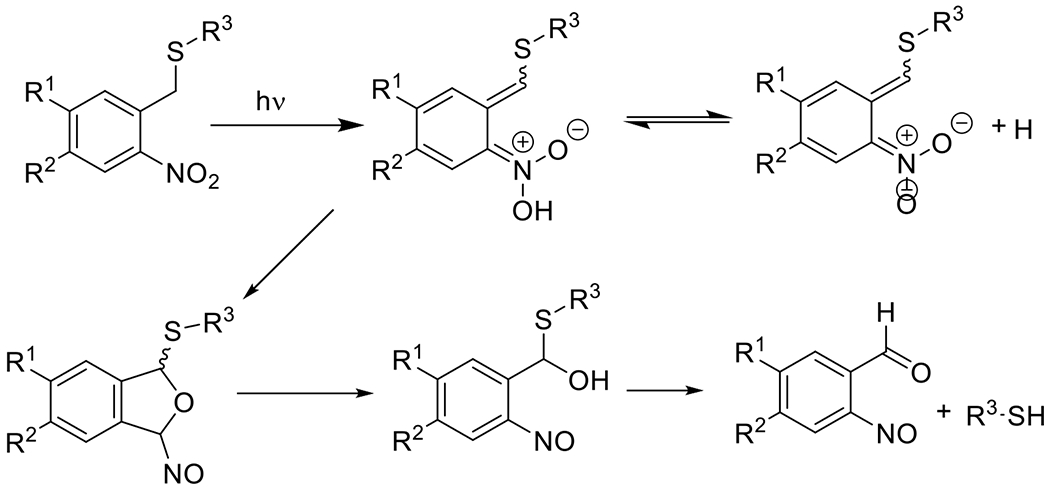
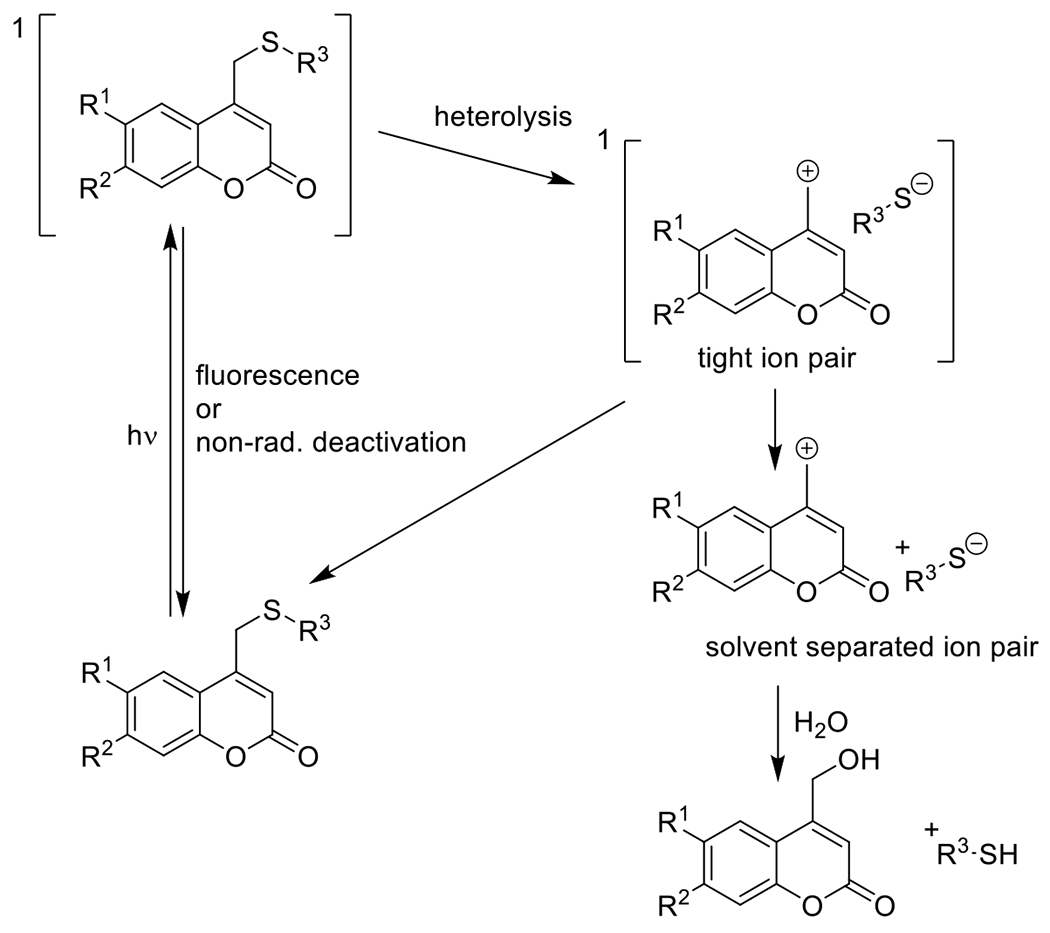
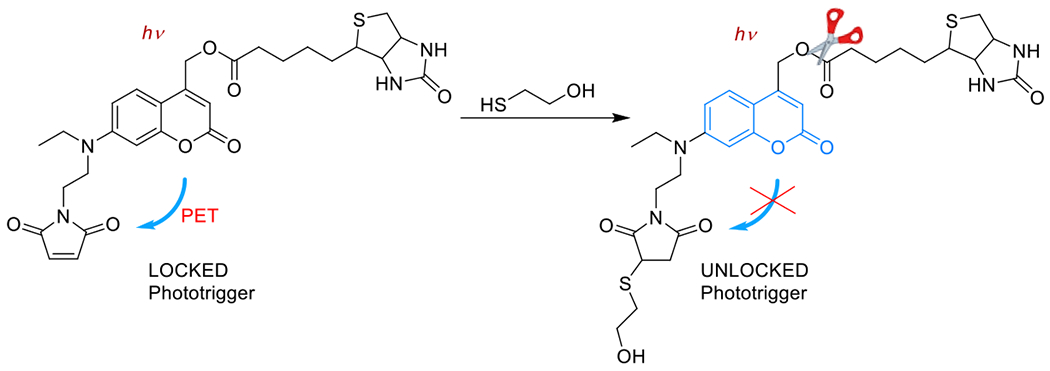
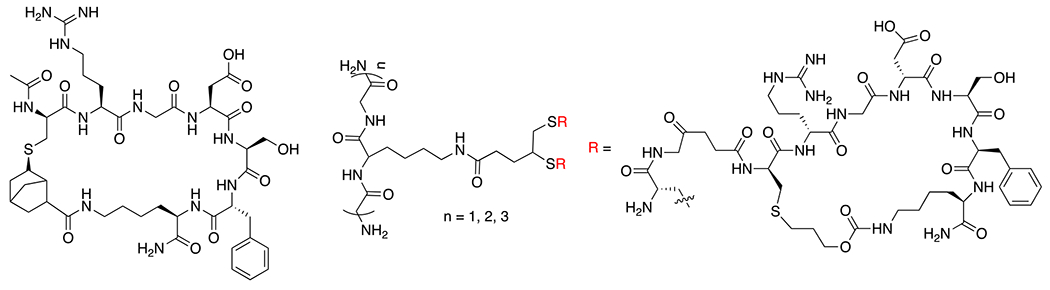

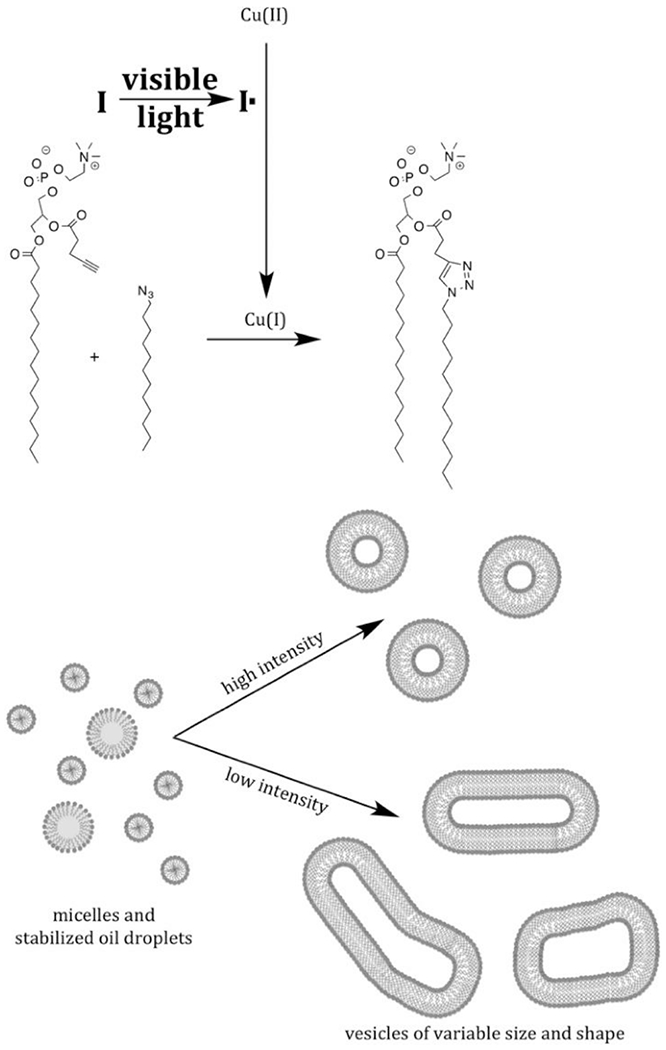
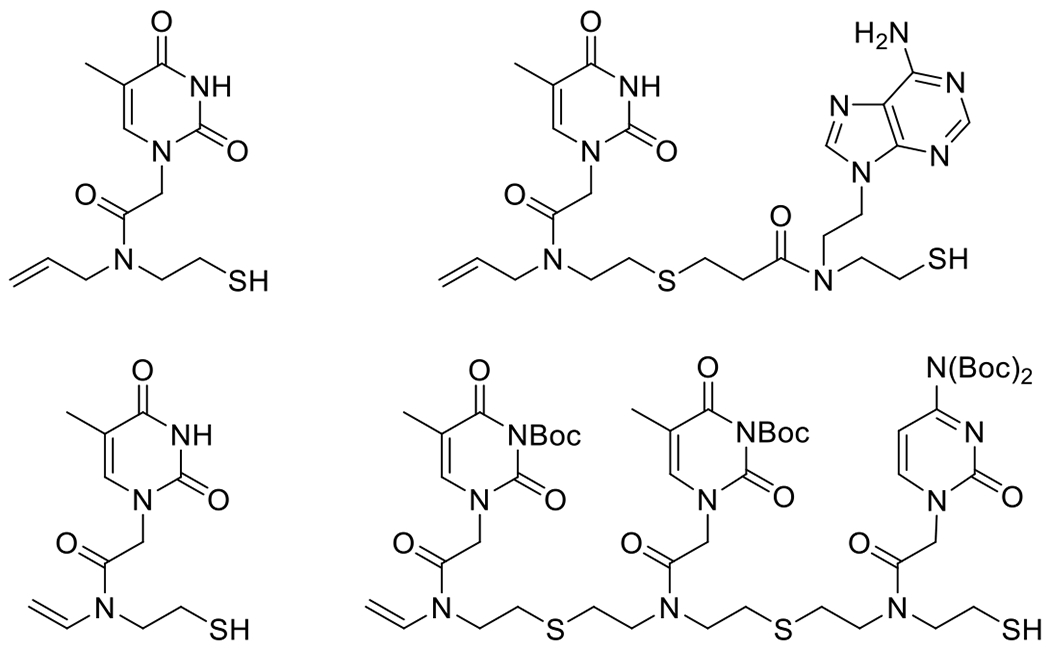
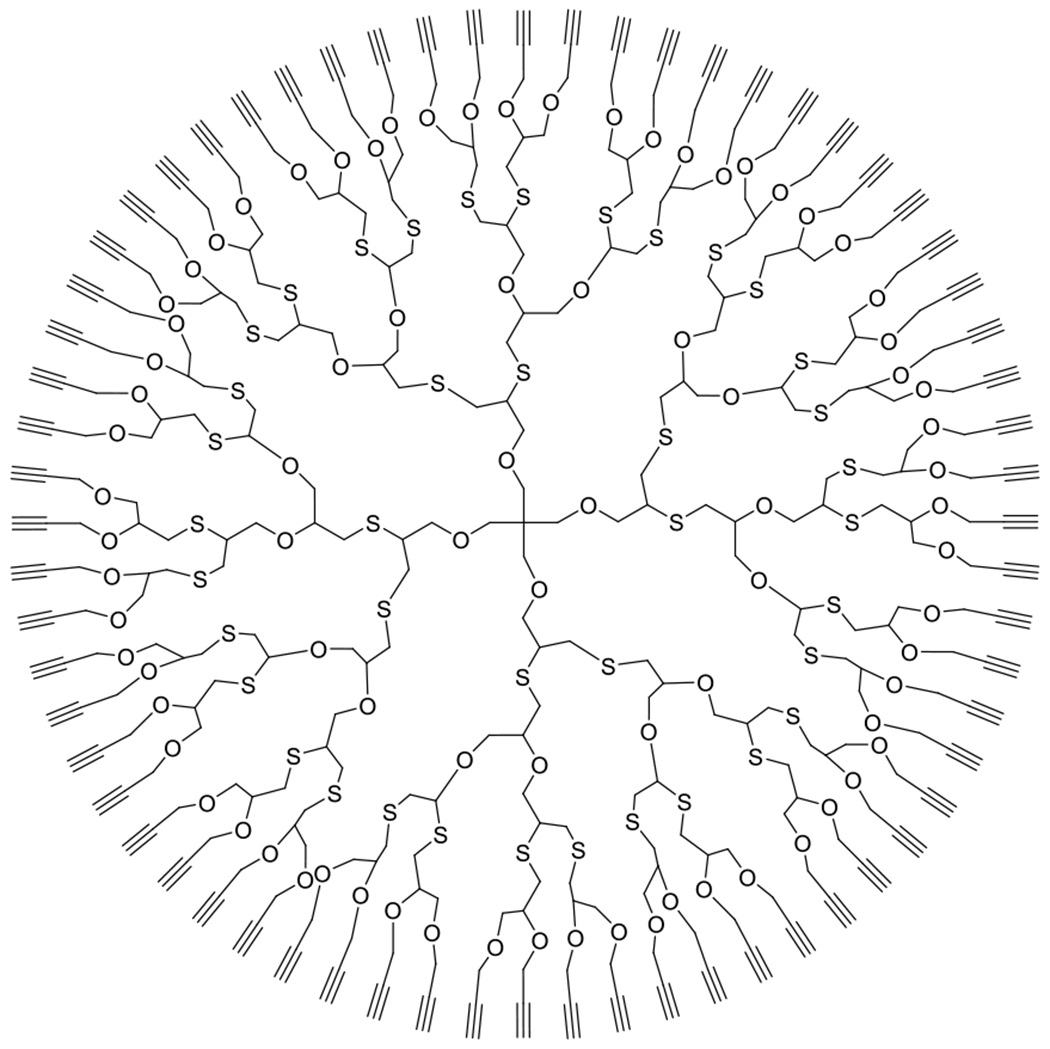


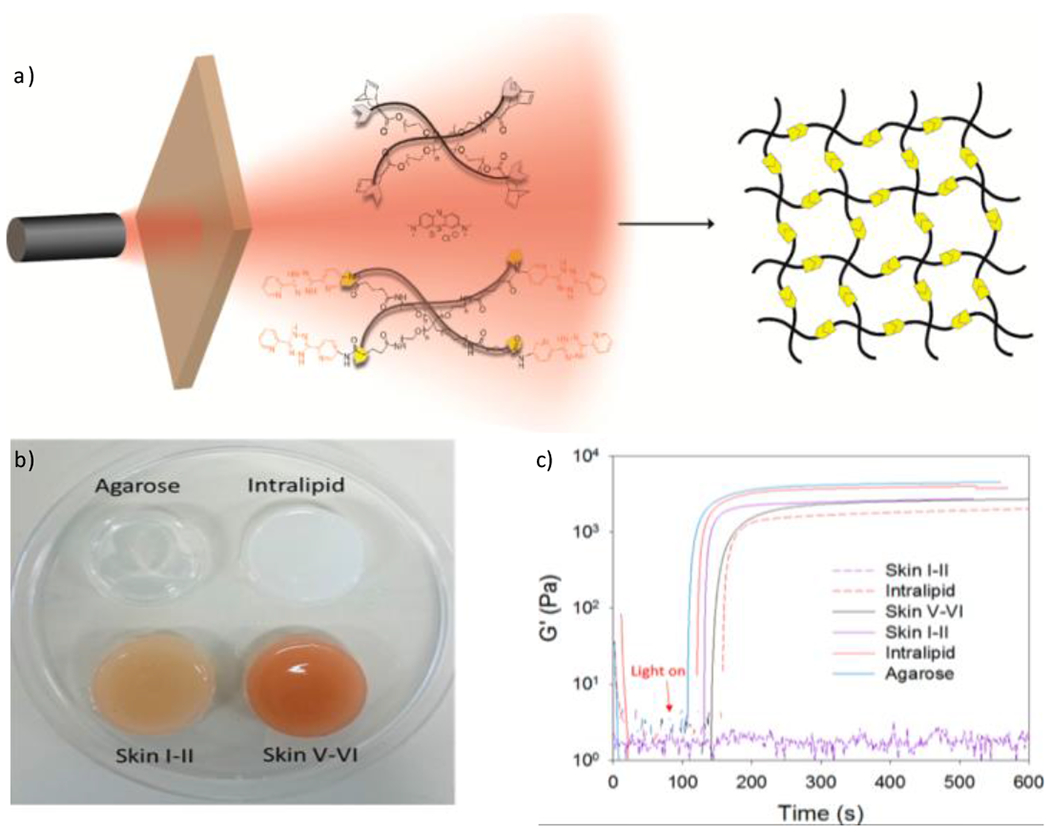
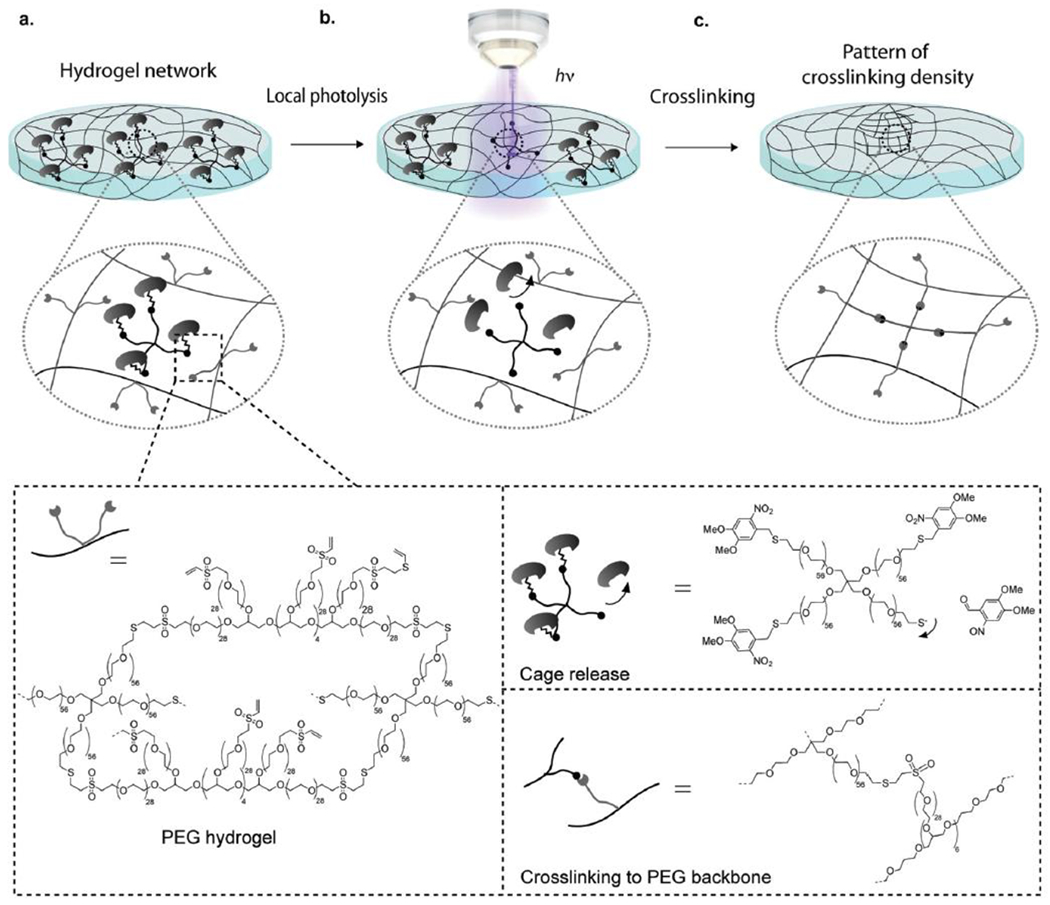
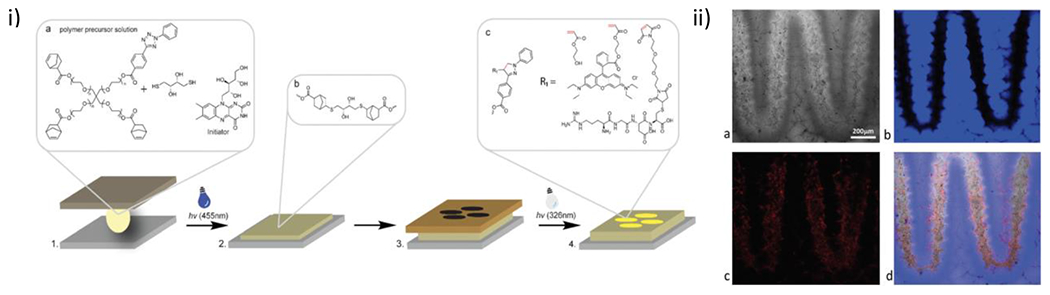
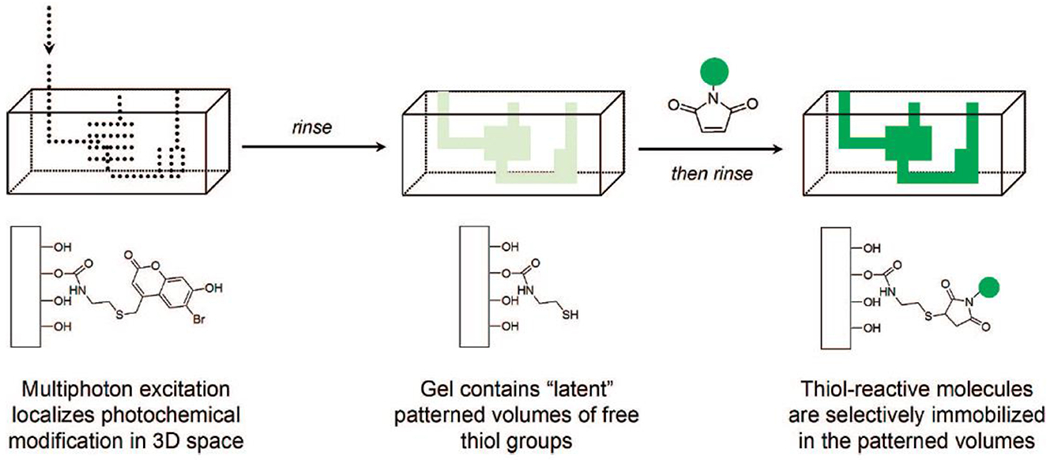
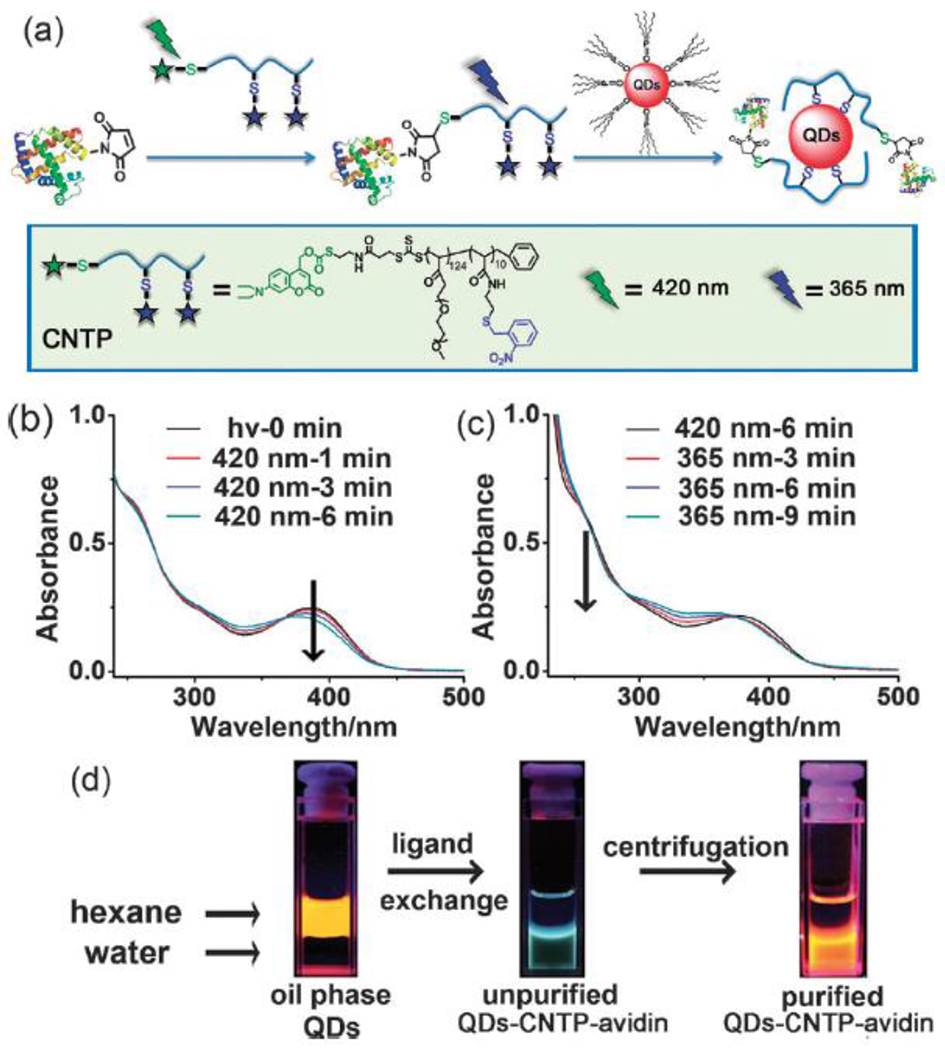
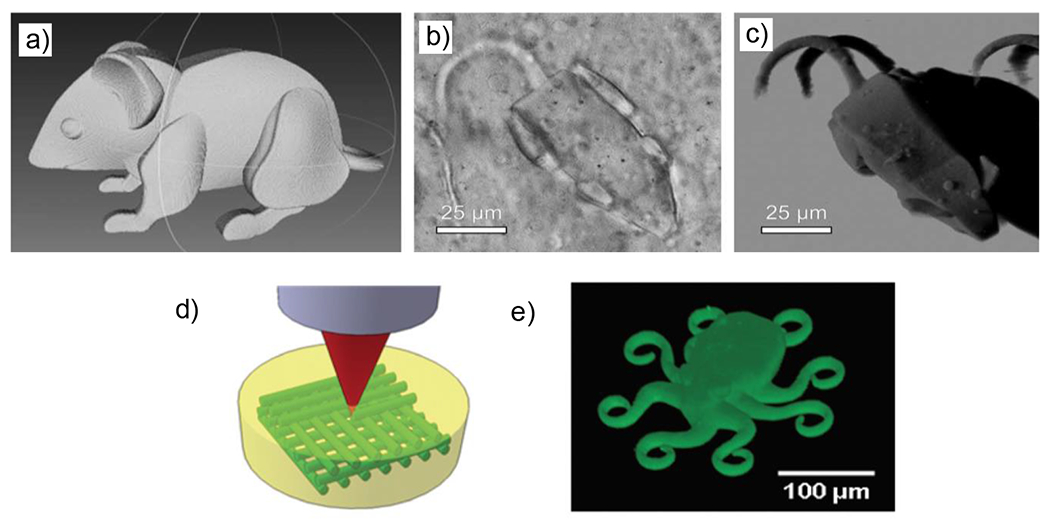
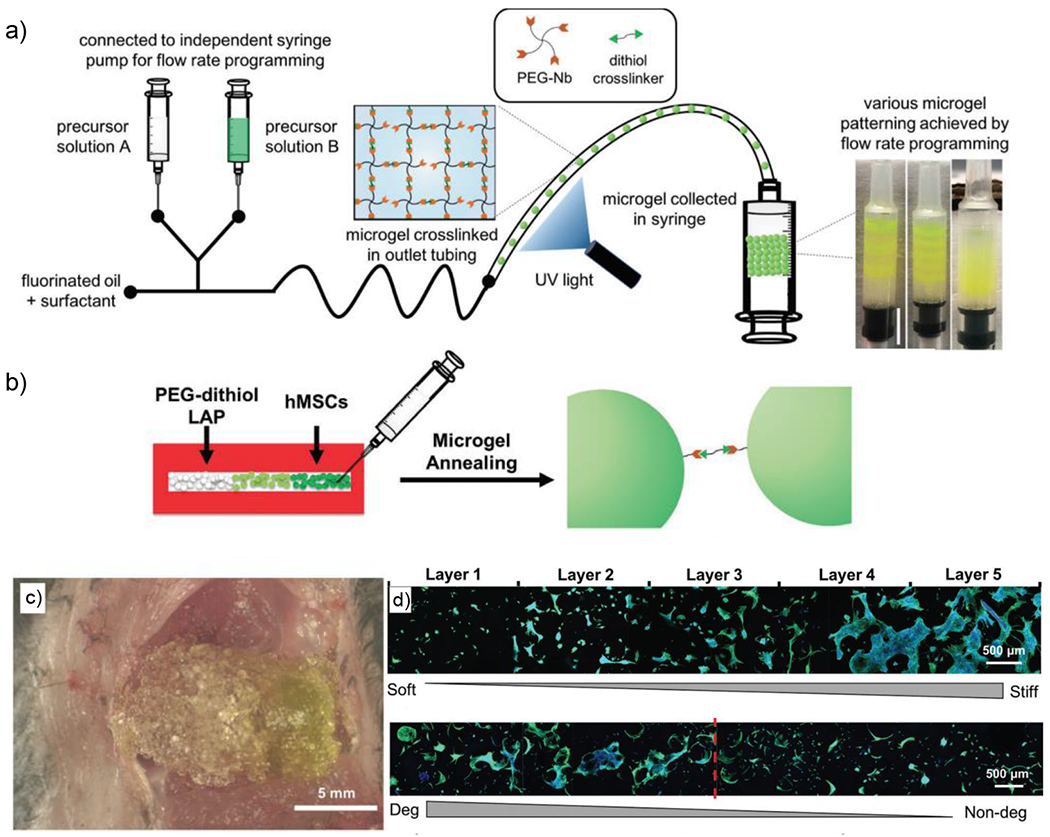
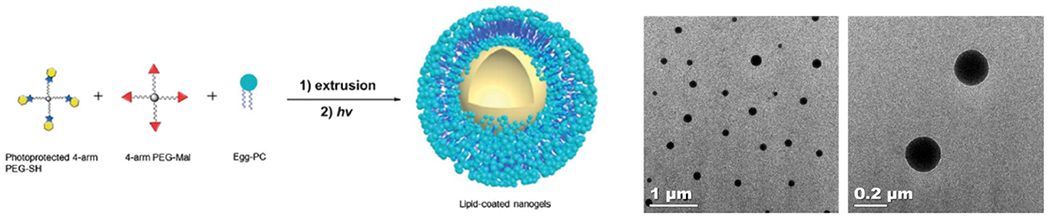
References
-
- Tasdelen MA and Yagci Y, Light-Induced Click Reactions, Angew. Chem. Int. Ed, 2013, 52, 5930–5938. - PubMed
-
- Kolb HC, Finn MG, and Sharpless KB, Click Chemistry: Diverse Chemical Function from a Few Good Reactions, Angew. Chem. Int. Ed, 2001, 40, 2004-+. - PubMed
-
- Barner-Kowollik C, Du Prez FE, Espeel P, Hawker CJ, Junkers T, Schlaad H, and Van Camp W, “Clicking” Polymers or Just Efficient Linking: What Is the Difference?, Angew. Chem. Int. Ed, 2011, 50, 60–62. - PubMed
-
- Fairbanks BD, Love DM, and Bowman CN, Efficient Polymer-Polymer Conjugation Via Thiol-Ene Click Reaction, Macromol. Chem. Phys, 2017, 218.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
