

Efficient mixed model approach for large-scale genome-wide association studies of ordinal categorical phenotypes
- PMID: 33836139
- PMCID: PMC8206161
- DOI: 10.1016/j.ajhg.2021.03.019
Efficient mixed model approach for large-scale genome-wide association studies of ordinal categorical phenotypes
Abstract
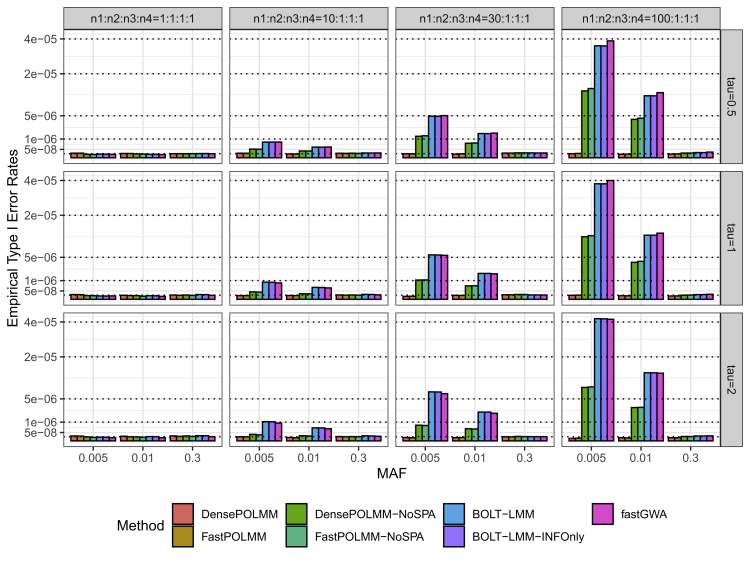
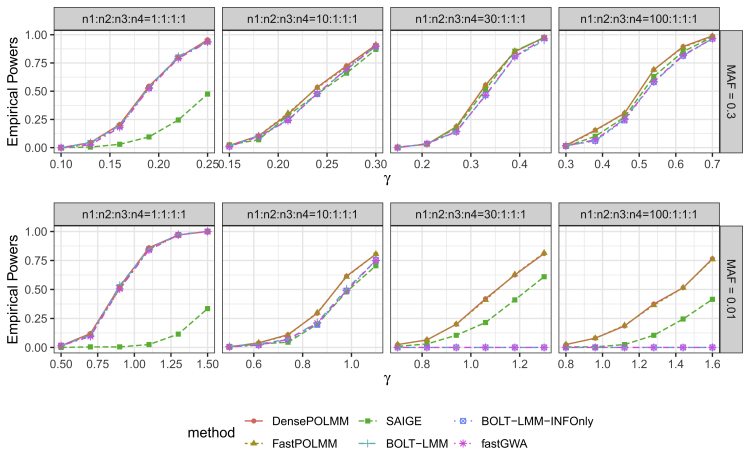
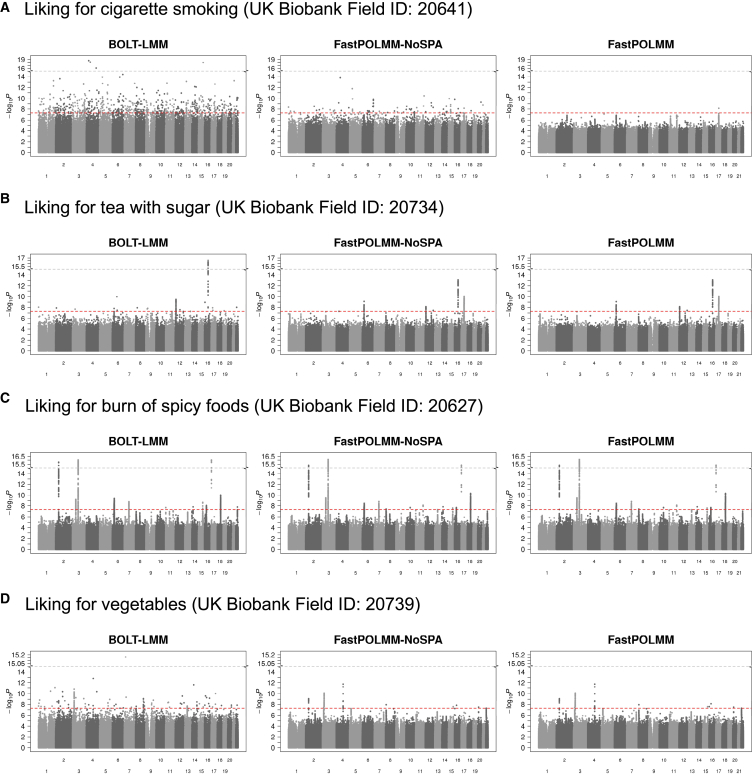
In genome-wide association studies, ordinal categorical phenotypes are widely used to measure human behaviors, satisfaction, and preferences. However, because of the lack of analysis tools, methods designed for binary or quantitative traits are commonly used inappropriately to analyze categorical phenotypes. To accurately model the dependence of an ordinal categorical phenotype on covariates, we propose an efficient mixed model association test, proportional odds logistic mixed model (POLMM). POLMM is computationally efficient to analyze large datasets with hundreds of thousands of samples, can control type I error rates at a stringent significance level regardless of the phenotypic distribution, and is more powerful than alternative methods. In contrast, the standard linear mixed model approaches cannot control type I error rates for rare variants when the phenotypic distribution is unbalanced, although they performed well when testing common variants. We applied POLMM to 258 ordinal categorical phenotypes on array genotypes and imputed samples from 408,961 individuals in UK Biobank. In total, we identified 5,885 genome-wide significant variants, of which, 424 variants (7.2%) are rare variants with MAF < 0.01.
Keywords: GRM; GWAS; POLMM; PheWAS; UK Biobank; food and other preferences; genetic relationship matrix; genome-wide association studies; mixed model approach; ordinal categorical data; phenome-wide association studies; proportional odds logistic mixed model; saddlepoint approximation; unbalanced phenotypic distribution.
Copyright © 2021 American Society of Human Genetics. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Beesley L.J., Salvatore M., Fritsche L.G., Pandit A., Rao A., Brummett C., Willer C.J., Lisabeth L.D., Mukherjee B. The emerging landscape of health research based on biobanks linked to electronic health records: Existing resources, statistical challenges, and potential opportunities. Stat. Med. 2019;39:773–800. - PMC - PubMed
-
- Agresti A. John Wiley & Sons; 2003. Categorical data analysis.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
