

IL-6 modulation for COVID-19: the right patients at the right time?
- PMID: 33837054
- PMCID: PMC8042594
- DOI: 10.1136/jitc-2020-002285
IL-6 modulation for COVID-19: the right patients at the right time?
Abstract
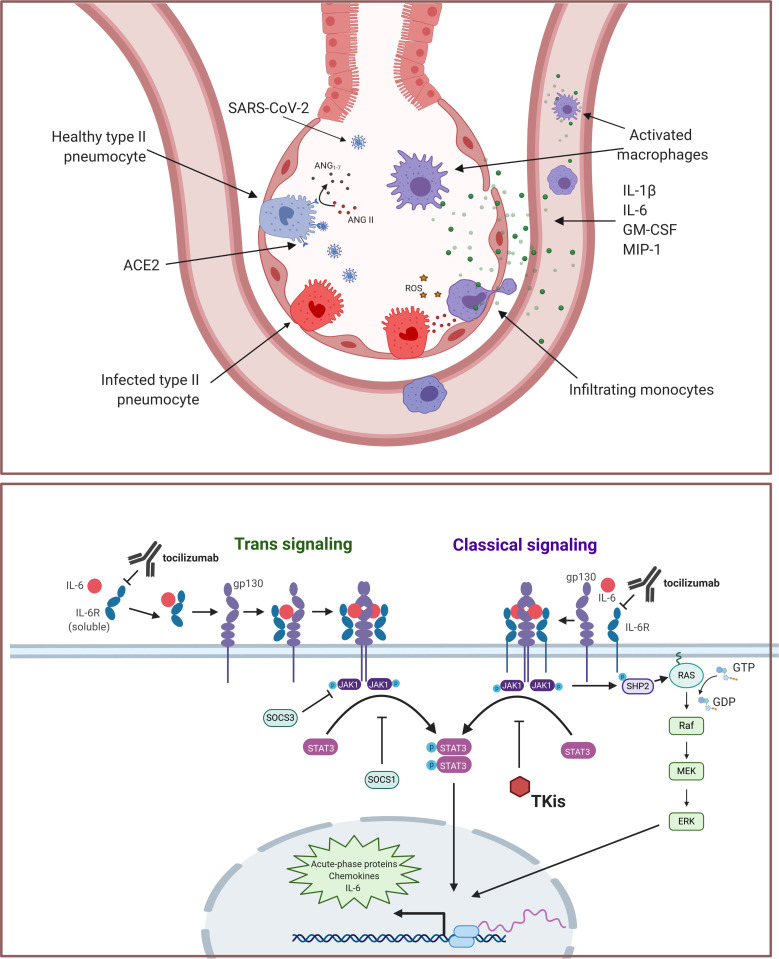
The ongoing pandemic caused by the novel coronavirus SARS-CoV-2 has disrupted the global economy and strained healthcare systems to their limits. After the virus first emerged in late 2019, the first intervention that demonstrated significant reductions in mortality for severe COVID-19 in large-scale trials was corticosteroids. Additional options that may reduce the burden on the healthcare system by reducing the number of patients requiring intensive care unit support are desperately needed, yet no therapy has conclusively established benefit in randomized studies for the management of moderate or mild cases of disease. Severe COVID-19 disease is characterized by a respiratory distress syndrome accompanied by elevated levels of several systemic cytokines, in a profile that shares several features with known inflammatory pathologies such as hemophagocytic lymphohistiocytosis and cytokine release syndrome secondary to chimeric antigen receptor (CAR) T cell therapy. Based on these observations, modulation of inflammatory cytokines, particularly interleukin (IL)-6, was proposed as a strategy to mitigate severe disease. Despite encouraging recoveries with anti-IL-6 agents, especially tocilizumab from single-arm studies, early randomized trials returned mixed results in terms of clinical benefit with these interventions. Later, larger trials such as RECOVERY and REMAP-CAP, however, are establishing anti-IL-6 in combination with steroids as a potential option for hypoxic patients with evidence of hyperinflammation. We propose that a positive feedback loop primarily mediated by macrophages and monocytes initiates the inflammatory cascade in severe COVID-19, and thus optimal benefit with anti-IL-6 therapies may require intervention during a finite window of opportunity at the outset of hyperinflammation but before fulminant disease causes irreversible tissue damage-as defined clinically by C reactive protein levels higher than 75 mg/L.
Keywords: COVID-19; cytokines.
© Author(s) (or their employer(s)) 2021. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: PAA has/had a consultant/advisory role for Bristol Myers Squibb, Roche-Genentech, Merck Sharp & Dohme, Novartis, Array, Merck Serono, Pierre-Fabre, Incyte, Medimmune, AstraZeneca, Syndax, Sun Pharma, Sanofi, Idera, Ultimovacs, Sandoz, Immunocore, 4SC, Alkermes, Italfarmaco, Nektar, Boehringer-Ingelheim, Eisai, Regeneron, Daiichi Sankyo, Pfizer, Oncosec, Nouscom, Takis, Lunaphore. He also received research funding from Bristol Myers Squibb, Roche-Genentech, Array, Sanofi and travel support from MSD. All the other authors have no conflict of interest to declare.
Figures
Similar articles
-
Treatment of severely ill COVID-19 patients with anti-interleukin drugs (COV-AID): A structured summary of a study protocol for a randomised controlled trial.Trials. 2020 Jun 3;21(1):468. doi: 10.1186/s13063-020-04453-5. Trials. 2020. PMID: 32493441 Free PMC article.
-
Clinical outcomes in COVID-19 patients treated with tocilizumab: An individual patient data systematic review.J Med Virol. 2020 Nov;92(11):2516-2522. doi: 10.1002/jmv.26038. Epub 2020 Jun 9. J Med Virol. 2020. PMID: 32436994 Free PMC article.
-
The Society for Immunotherapy of Cancer perspective on regulation of interleukin-6 signaling in COVID-19-related systemic inflammatory response.J Immunother Cancer. 2020 May;8(1):e000930. doi: 10.1136/jitc-2020-000930. J Immunother Cancer. 2020. PMID: 32385146 Free PMC article.
-
Hemophagocytic lymphohistiocytosis in a patient with COVID-19 treated with tocilizumab: a case report.J Med Case Rep. 2020 Oct 15;14(1):187. doi: 10.1186/s13256-020-02503-9. J Med Case Rep. 2020. PMID: 33054818 Free PMC article.
-
Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)?J Autoimmun. 2020 Jul;111:102452. doi: 10.1016/j.jaut.2020.102452. Epub 2020 Apr 10. J Autoimmun. 2020. PMID: 32291137 Free PMC article. Review.
Cited by
-
Vitamin B12 attenuates leukocyte inflammatory signature in COVID-19 via methyl-dependent changes in epigenetic markings.Front Immunol. 2023 Mar 13;14:1048790. doi: 10.3389/fimmu.2023.1048790. eCollection 2023. Front Immunol. 2023. PMID: 36993968 Free PMC article.
-
Do All Critically Ill Patients with COVID-19 Disease Benefit from Adding Tocilizumab to Glucocorticoids? A Retrospective Cohort Study.Viruses. 2023 Jan 20;15(2):294. doi: 10.3390/v15020294. Viruses. 2023. PMID: 36851508 Free PMC article.
-
An updated overview of recent advances, challenges, and clinical considerations of IL-6 signaling blockade in severe coronavirus disease 2019 (COVID-19).Int Immunopharmacol. 2022 Apr;105:108536. doi: 10.1016/j.intimp.2022.108536. Epub 2022 Jan 11. Int Immunopharmacol. 2022. PMID: 35074571 Free PMC article. Review.
-
Interleukin-6 in SARS-CoV-2 induced disease: Interactions and therapeutic applications.Biomed Pharmacother. 2022 Jan;145:112419. doi: 10.1016/j.biopha.2021.112419. Epub 2021 Nov 12. Biomed Pharmacother. 2022. PMID: 34781146 Free PMC article. Review.
-
Cancer bio-immunotherapy XVIII annual NIBIT-(Italian network for tumor biotherapy) meeting, October 15-16, 2020.Cancer Immunol Immunother. 2022 Jul;71(7):1787-1794. doi: 10.1007/s00262-022-03145-0. Epub 2022 Jan 16. Cancer Immunol Immunother. 2022. PMID: 35034143 Free PMC article. No abstract available.
References
-
- JohnsHopkins . Mortality analyses - Johns Hopkins Coronavirus resource center: JohnsHopkins, 2020. Available: https://coronavirus.jhu.edu/data/mortality
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous