

Mapping the regulatory landscape of auditory hair cells from single-cell multi-omics data
- PMID: 33837132
- PMCID: PMC8494220
- DOI: 10.1101/gr.271080.120
Mapping the regulatory landscape of auditory hair cells from single-cell multi-omics data
Abstract
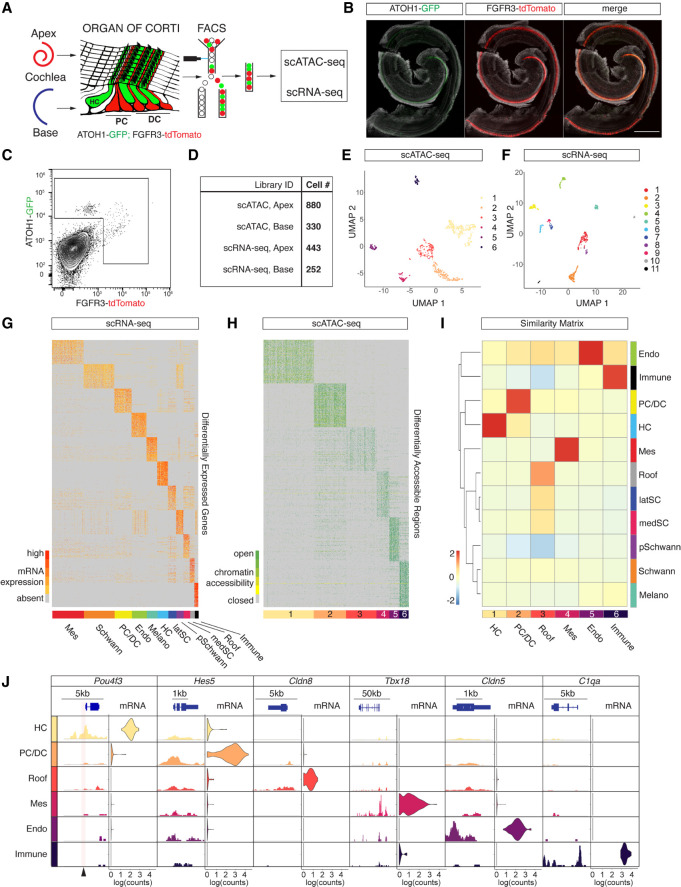
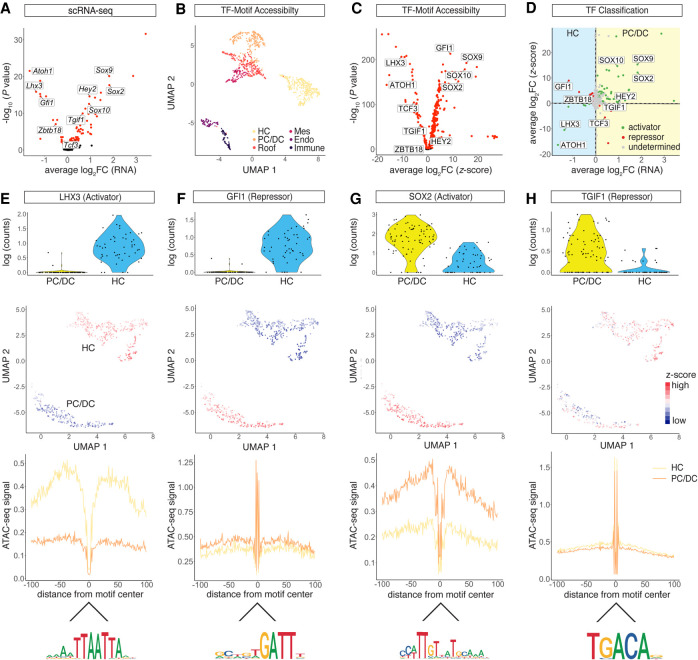
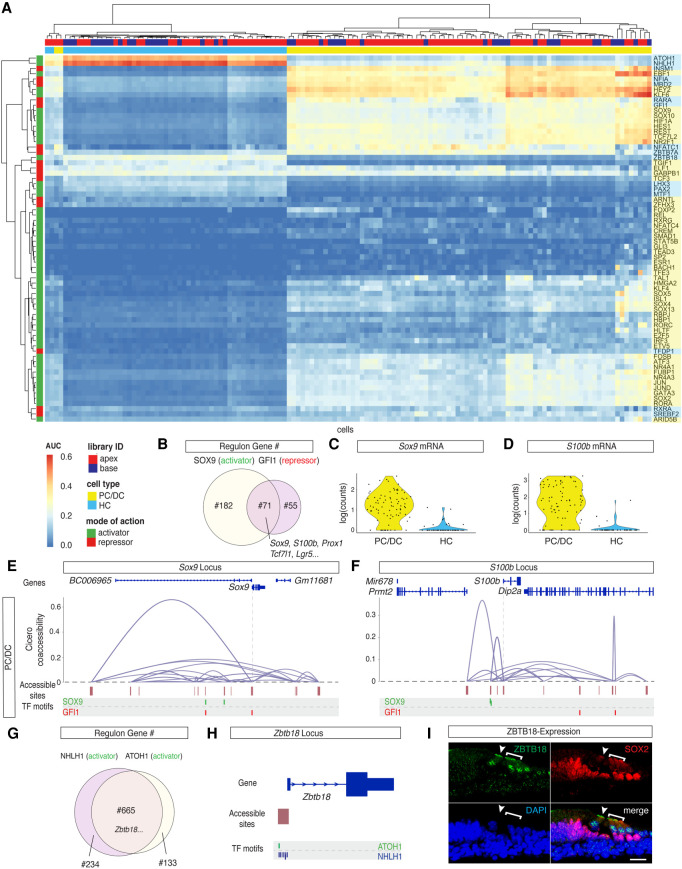
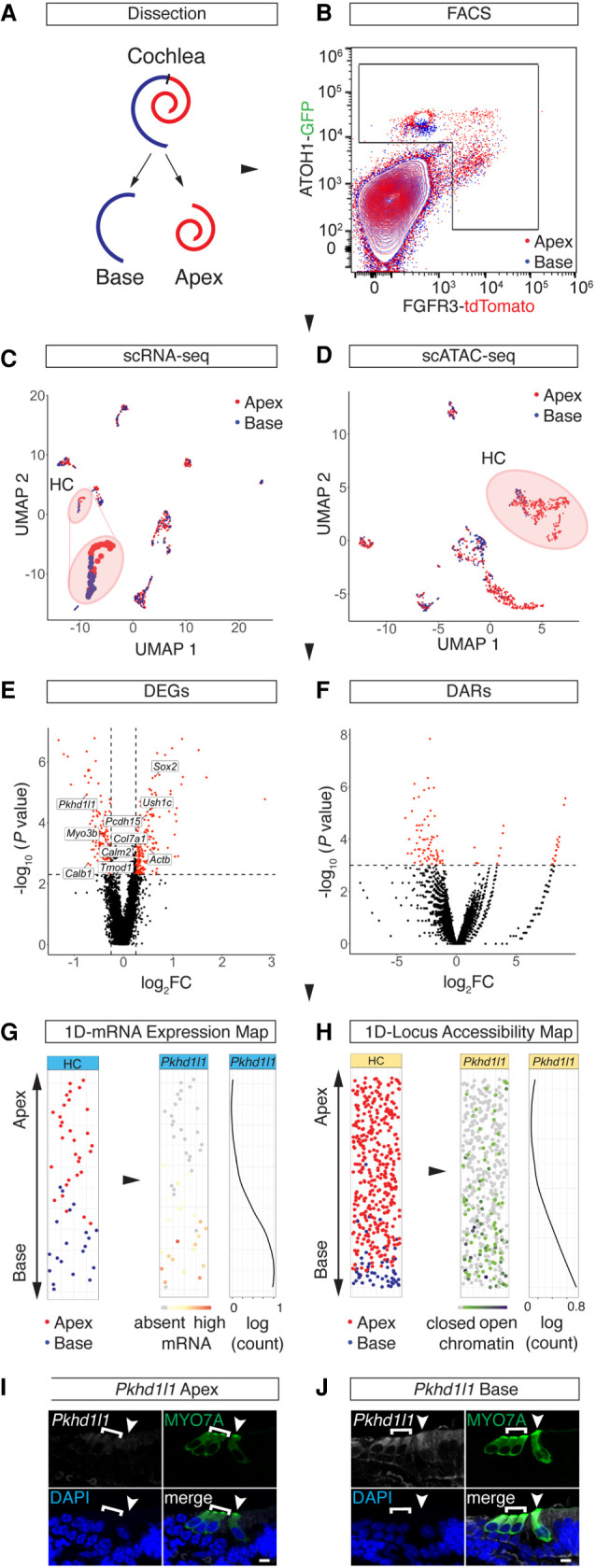
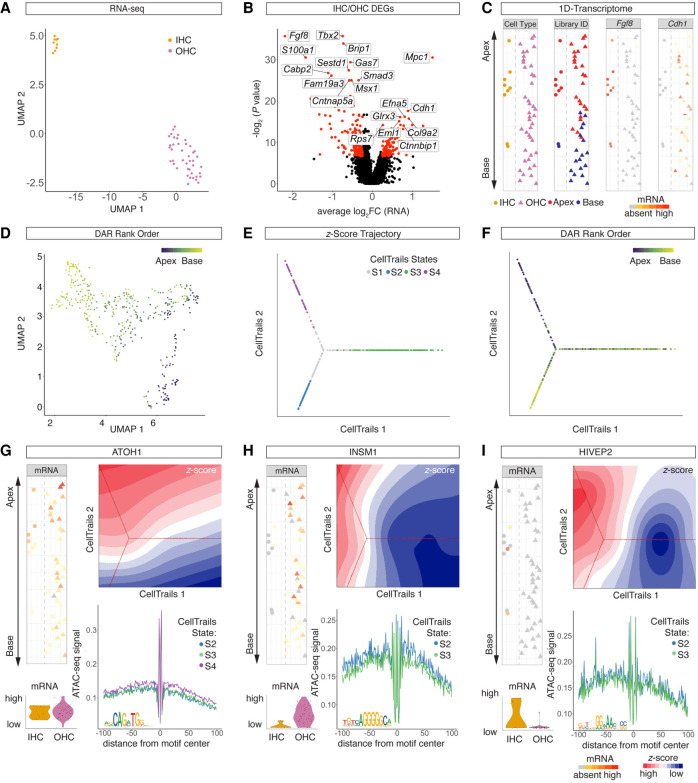
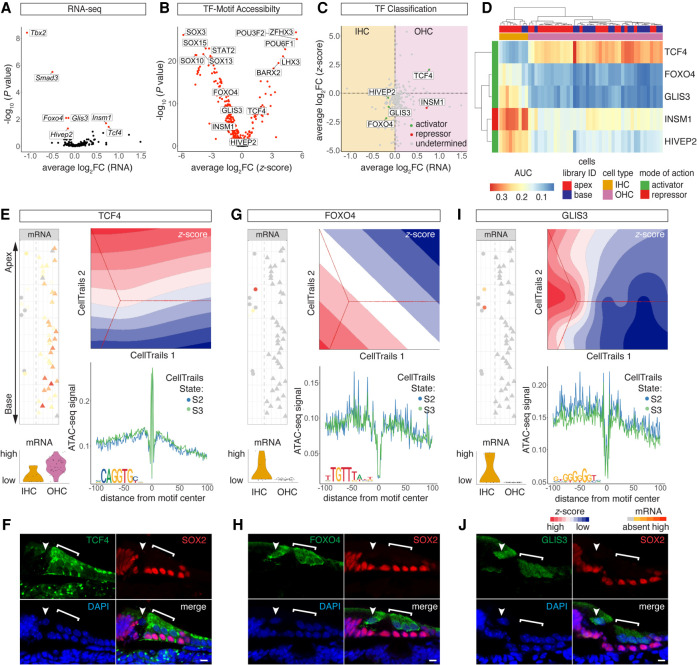
Auditory hair cells transduce sound to the brain, and in mammals, these cells reside together with supporting cells in the sensory epithelium of the cochlea, called the organ of Corti. To establish the organ's delicate function during development and differentiation, spatiotemporal gene expression is strictly controlled by chromatin accessibility and cell type-specific transcription factors, jointly representing the regulatory landscape. Bulk sequencing technology and cellular heterogeneity obscured investigations on the interplay between transcription factors and chromatin accessibility in inner ear development. To study the formation of the regulatory landscape in hair cells, we collected single-cell chromatin accessibility profiles accompanied by single-cell RNA data from genetically labeled murine hair cells and supporting cells after birth. Using an integrative approach, we predicted cell type-specific activating and repressing functions of developmental transcription factors. Furthermore, by integrating gene expression and chromatin accessibility data sets, we reconstructed gene regulatory networks. Then, using a comparative approach, 20 hair cell-specific activators and repressors, including putative downstream target genes, were identified. Clustering of target genes resolved groups of related transcription factors and was used to infer their developmental functions. Finally, the heterogeneity in the single-cell data allowed us to spatially reconstruct transcriptional as well as chromatin accessibility trajectories, indicating that gradual changes in the chromatin accessibility landscape are lagging behind the transcriptional identity of hair cells along the organ's longitudinal axis. Overall, this study provides a strategy to spatially reconstruct the formation of a lineage-specific regulatory landscape using a single-cell multi-omics approach.
© 2021 Wang et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
SoxC transcription factors shape the epigenetic landscape to establish competence for sensory differentiation in the mammalian organ of Corti.Proc Natl Acad Sci U S A. 2023 Aug 22;120(34):e2301301120. doi: 10.1073/pnas.2301301120. Epub 2023 Aug 16. Proc Natl Acad Sci U S A. 2023. PMID: 37585469 Free PMC article.
-
Open chromatin dynamics in prosensory cells of the embryonic mouse cochlea.Sci Rep. 2019 Jun 21;9(1):9060. doi: 10.1038/s41598-019-45515-2. Sci Rep. 2019. PMID: 31227770 Free PMC article.
-
Cochlear organoids reveal transcriptional programs of postnatal hair cell differentiation from supporting cells.Cell Rep. 2023 Nov 28;42(11):113421. doi: 10.1016/j.celrep.2023.113421. Epub 2023 Nov 11. Cell Rep. 2023. PMID: 37952154 Free PMC article.
-
Atoh1 and other related key regulators in the development of auditory sensory epithelium in the mammalian inner ear: function and interplay.Dev Biol. 2019 Feb 15;446(2):133-141. doi: 10.1016/j.ydbio.2018.12.025. Epub 2018 Dec 31. Dev Biol. 2019. PMID: 30605626 Review.
-
Development in the Mammalian Auditory System Depends on Transcription Factors.Int J Mol Sci. 2021 Apr 18;22(8):4189. doi: 10.3390/ijms22084189. Int J Mol Sci. 2021. PMID: 33919542 Free PMC article. Review.
Cited by
-
Defining the identity and the niches of epithelial stem cells with highly pleiotropic multilineage potency in the human thymus.Dev Cell. 2023 Nov 20;58(22):2428-2446.e9. doi: 10.1016/j.devcel.2023.08.017. Epub 2023 Aug 30. Dev Cell. 2023. PMID: 37652013 Free PMC article.
-
Mapping the developmental potential of mouse inner ear organoids at single-cell resolution.iScience. 2024 Feb 1;27(3):109069. doi: 10.1016/j.isci.2024.109069. eCollection 2024 Mar 15. iScience. 2024. PMID: 38375227 Free PMC article.
-
Single-Cell Integration of BMD GWAS Results Prioritize Candidate Genes Influencing Age-Related Bone Loss.JBMR Plus. 2023 Jul 7;7(10):e10795. doi: 10.1002/jbm4.10795. eCollection 2023 Oct. JBMR Plus. 2023. PMID: 37808401 Free PMC article.
-
Developing a general AI model for integrating diverse genomic modalities and comprehensive genomic knowledge.bioRxiv [Preprint]. 2025 May 14:2025.05.08.652986. doi: 10.1101/2025.05.08.652986. bioRxiv. 2025. PMID: 40462903 Free PMC article. Preprint.
-
SOX9 and SOX10 control fluid homeostasis in the inner ear for hearing through independent and cooperative mechanisms.Proc Natl Acad Sci U S A. 2022 Nov 16;119(46):e2122121119. doi: 10.1073/pnas.2122121119. Epub 2022 Nov 7. Proc Natl Acad Sci U S A. 2022. PMID: 36343245 Free PMC article.
References
-
- Aleksiūnienė B, Matulevičiūtė R, Matulevičienė A, Burnytė B, Krasovskaja N, Ambrozaitytė L, Mikštienė V, Dirsė V, Utkus A, Kučinskas V. 2017. Opposite chromosome constitutions due to a familial translocation t(1;21)(q43;q22) in two cousins with development delay and congenital anomalies: a case report. Medicine (Baltimore) 96: e6521. 10.1097/MD.0000000000006521 - DOI - PMC - PubMed
-
- Berest I, Arnold C, Reyes-Palomares A, Palla G, Rasmussen KD, Giles H, Bruch PM, Huber W, Dietrich S, Helin K, et al. 2019. Quantification of differential transcription factor activity and multiomics-based classification into activators and repressors: diffTF. Cell Rep 29: 3147–3159.e12. 10.1016/j.celrep.2019.10.106 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases