

Oncogenic KRAS Recruits an Expansive Transcriptional Network through Mutant p53 to Drive Pancreatic Cancer Metastasis
- PMID: 33839689
- PMCID: PMC8338884
- DOI: 10.1158/2159-8290.CD-20-1228
Oncogenic KRAS Recruits an Expansive Transcriptional Network through Mutant p53 to Drive Pancreatic Cancer Metastasis
Abstract
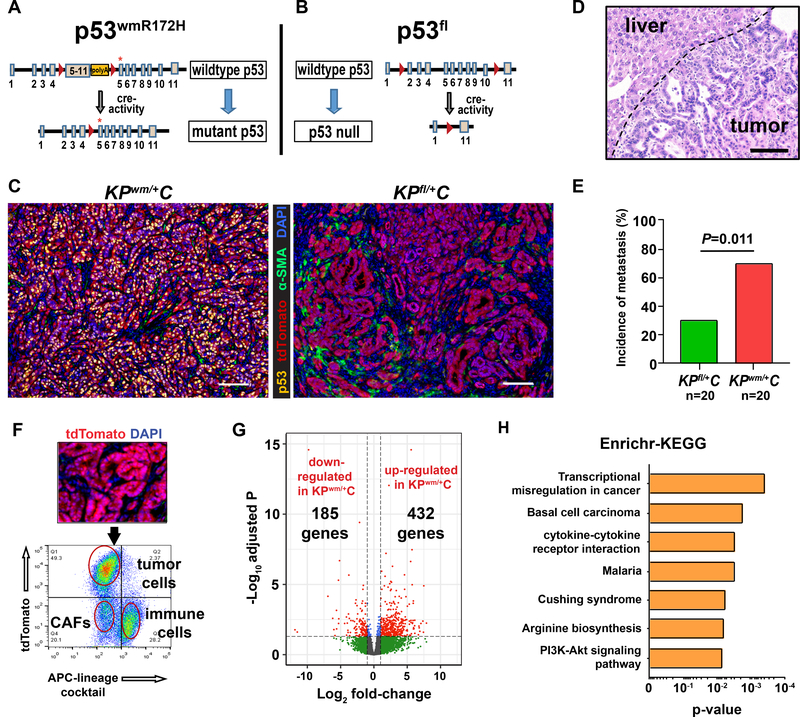
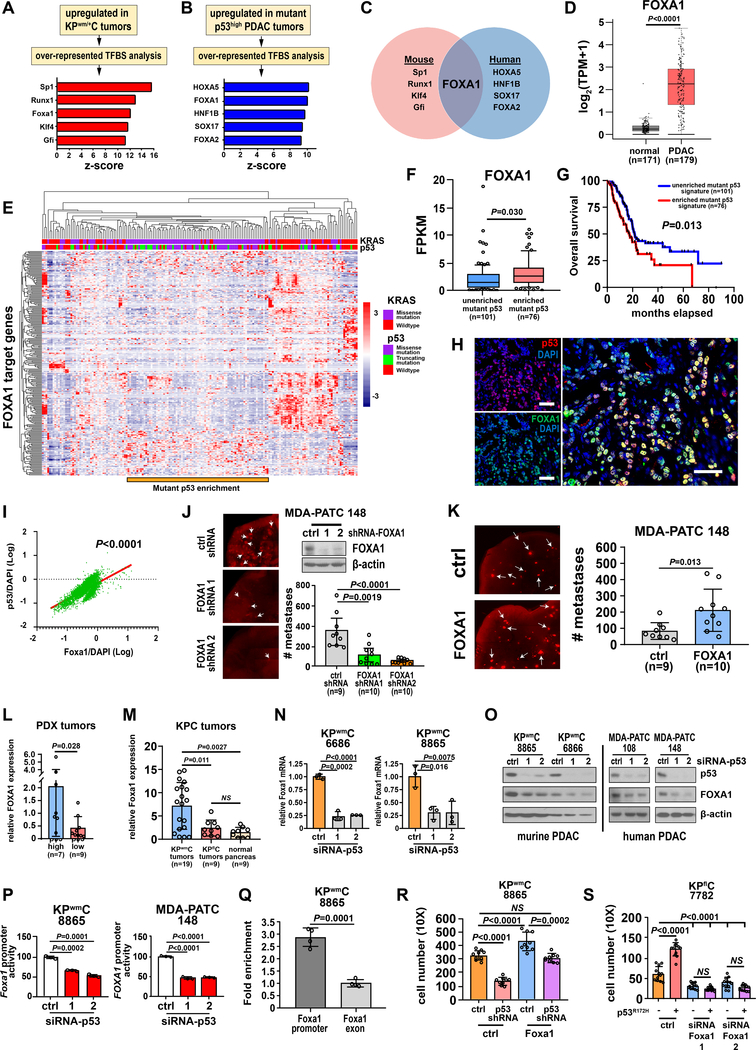
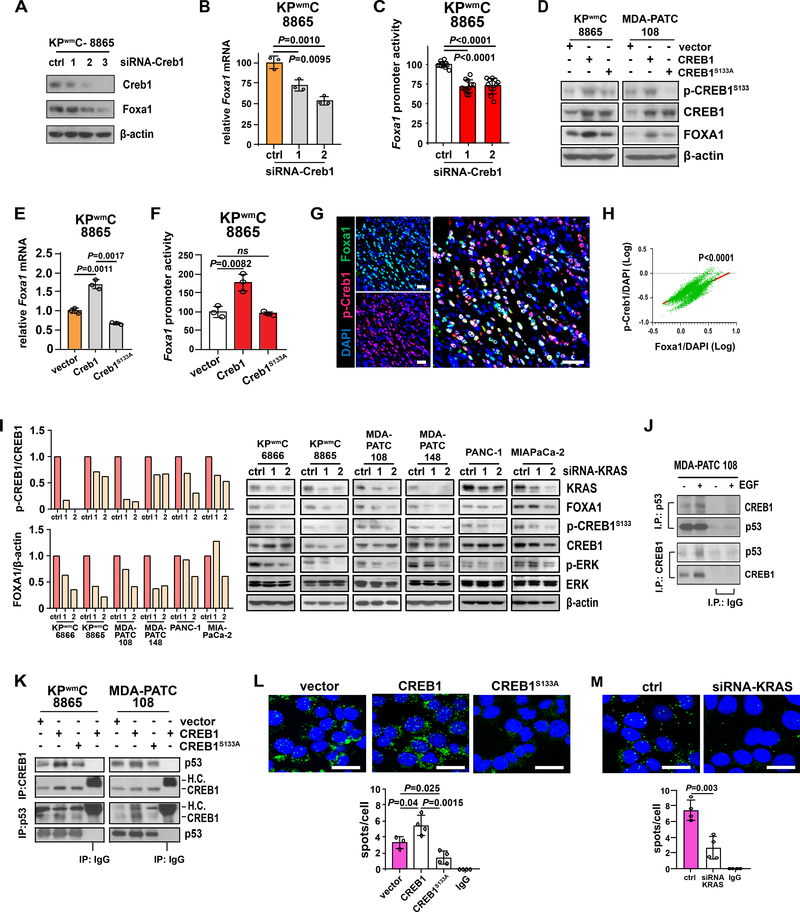
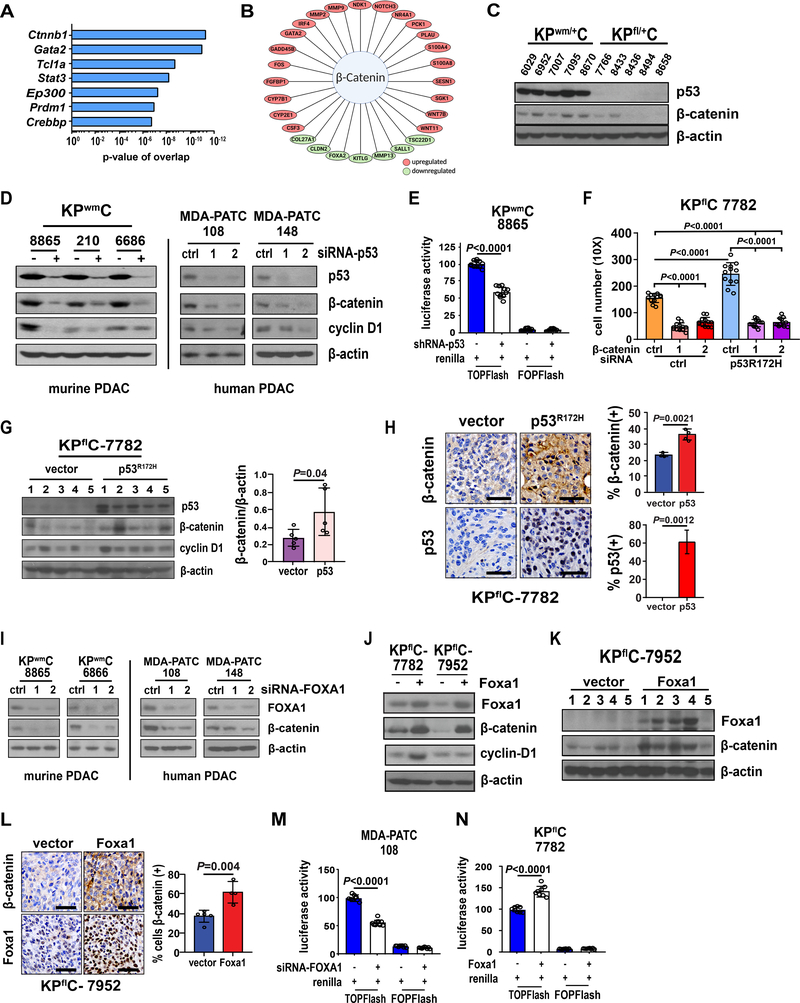
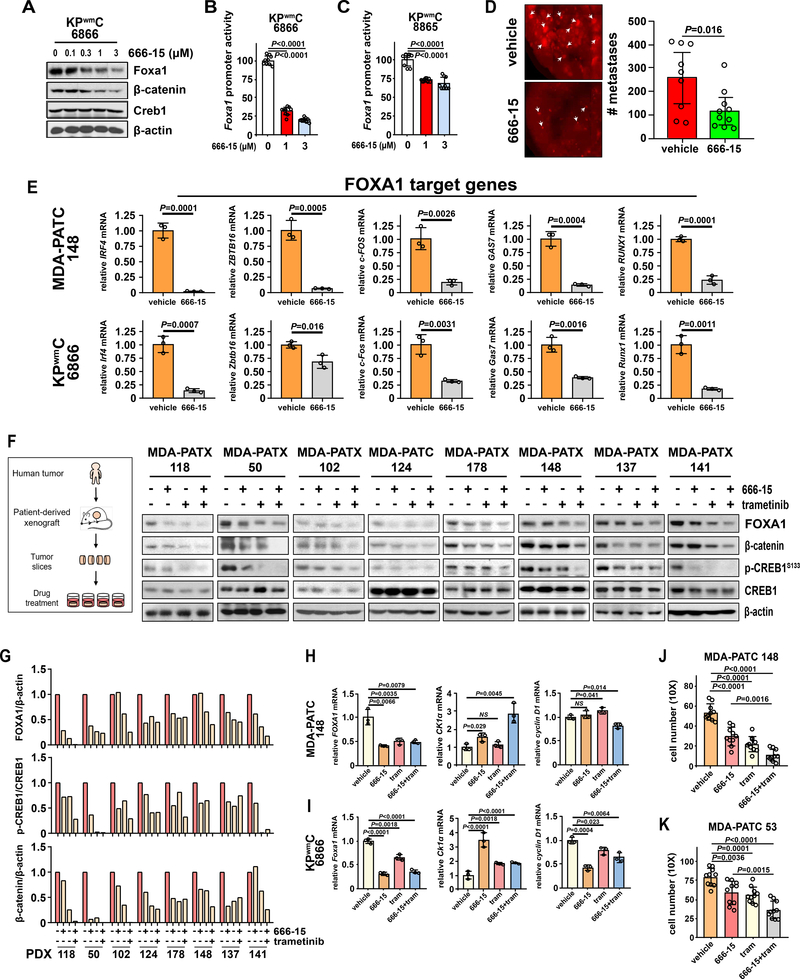
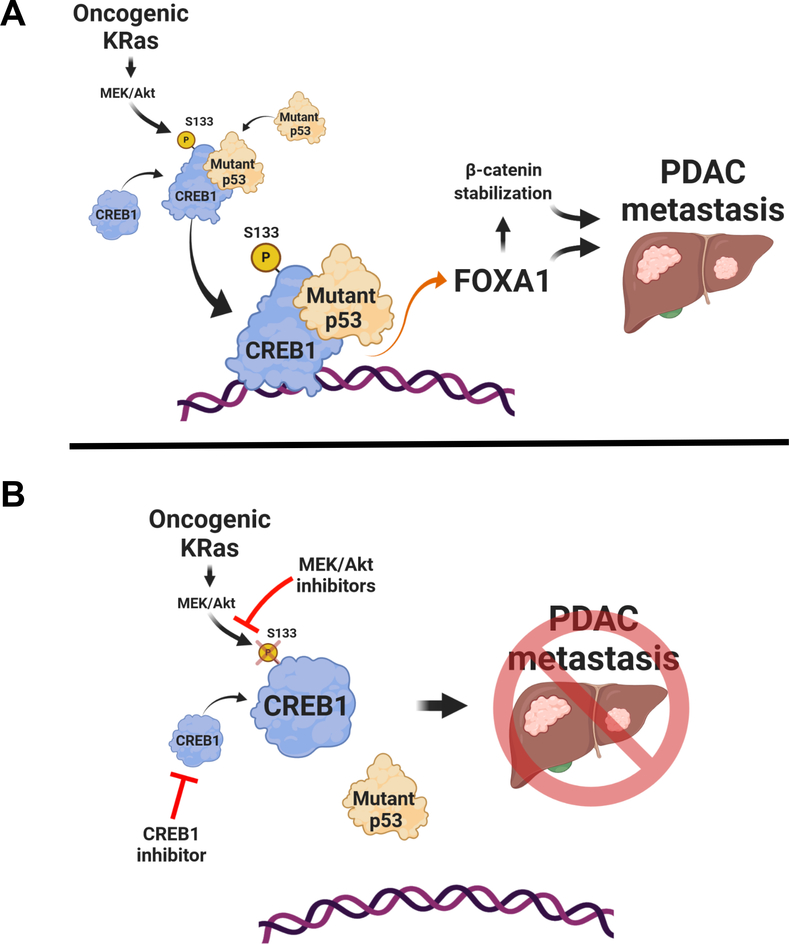
Pancreatic ductal adenocarcinoma (PDAC) is almost uniformly fatal and characterized by early metastasis. Oncogenic KRAS mutations prevail in 95% of PDAC tumors and co-occur with genetic alterations in the TP53 tumor suppressor in nearly 70% of patients. Most TP53 alterations are missense mutations that exhibit gain-of-function phenotypes that include increased invasiveness and metastasis, yet the extent of direct cooperation between KRAS effectors and mutant p53 remains largely undefined. We show that oncogenic KRAS effectors activate CREB1 to allow physical interactions with mutant p53 that hyperactivate multiple prometastatic transcriptional networks. Specifically, mutant p53 and CREB1 upregulate the prometastatic, pioneer transcription factor FOXA1, activating its transcriptional network while promoting WNT/β-catenin signaling, together driving PDAC metastasis. Pharmacologic CREB1 inhibition dramatically reduced FOXA1 and β-catenin expression and dampened PDAC metastasis, identifying a new therapeutic strategy to disrupt cooperation between oncogenic KRAS and mutant p53 to mitigate metastasis. SIGNIFICANCE: Oncogenic KRAS and mutant p53 are the most commonly mutated oncogene and tumor suppressor gene in human cancers, yet direct interactions between these genetic drivers remain undefined. We identified a cooperative node between oncogenic KRAS effectors and mutant p53 that can be therapeutically targeted to undermine cooperation and mitigate metastasis.This article is highlighted in the In This Issue feature, p. 1861.
©2021 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4(6):437–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
