

Alternate primers for whole-genome SARS-CoV-2 sequencing
- PMID: 33841912
- PMCID: PMC7928614
- DOI: 10.1093/ve/veab006
Alternate primers for whole-genome SARS-CoV-2 sequencing
Abstract
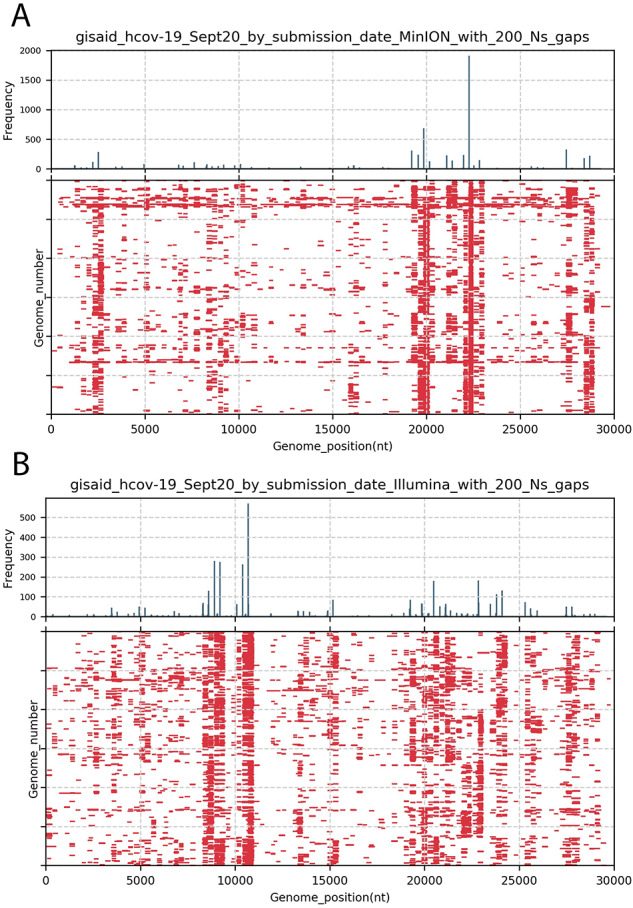
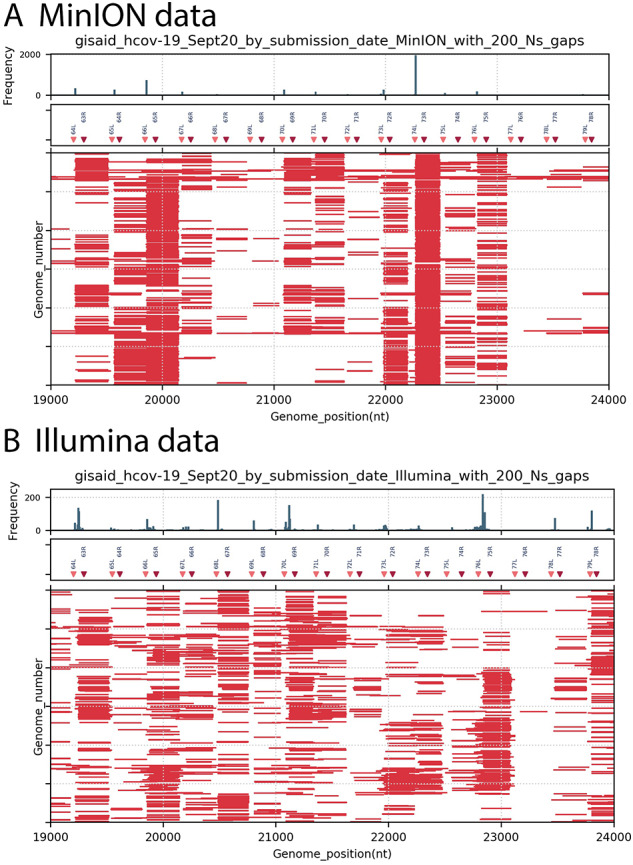
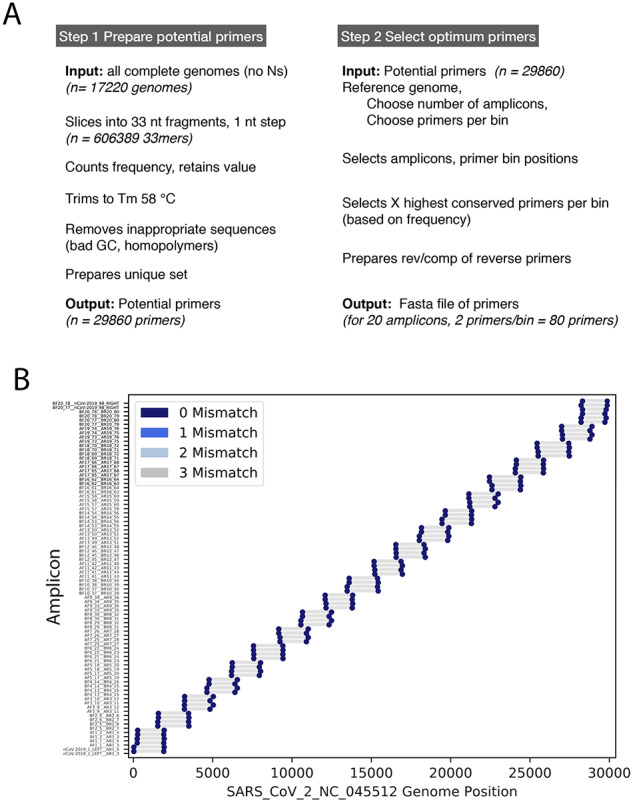
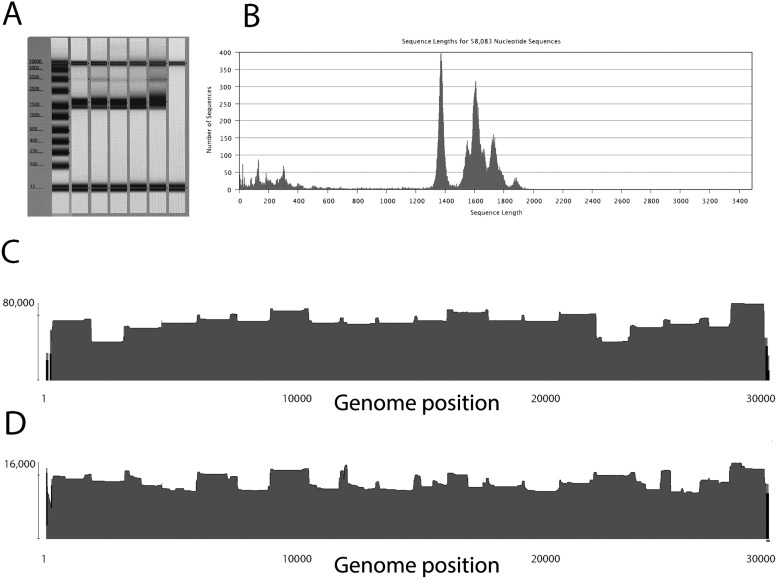
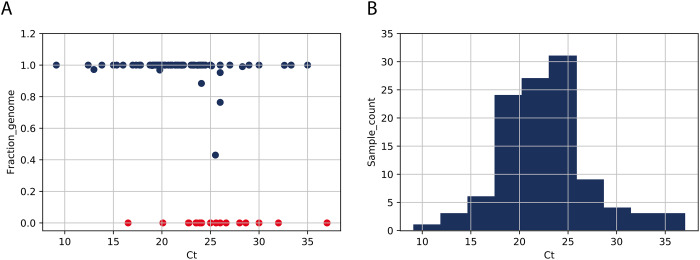
As the world is struggling to control the novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), there is an urgency to develop effective control measures. Essential information is encoded in the virus genome sequence with accurate and complete SARS-CoV-2 sequences essential for tracking the movement and evolution of the virus and for guiding efforts to develop vaccines and antiviral drugs. While there is unprecedented SARS-CoV-2 sequencing efforts globally, approximately 19 to 43 per cent of the genomes generated monthly are gapped, reducing their information content. The current study documents the genome gap frequencies and their positions in the currently available data and provides an alternative primer set and a sequencing scheme to help improve the quality and coverage of the genomes.
Keywords: COVID-19; SARS-CoV-2; next generation sequencing; primers.
© The Author(s) 2021. Published by Oxford University Press.
Figures
References
-
- De Maio N. et al. 2020. ‘Issues with SARS-CoV-2 Sequencing Data.’ <https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473> accessed 26 Jan 2021.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
