

Computational screening of FDA approved drugs of fungal origin that may interfere with SARS-CoV-2 spike protein activation, viral RNA replication, and post-translational modification: a multiple target approach
- PMID: 33842191
- PMCID: PMC8019482
- DOI: 10.1007/s40203-021-00089-8
Computational screening of FDA approved drugs of fungal origin that may interfere with SARS-CoV-2 spike protein activation, viral RNA replication, and post-translational modification: a multiple target approach
Abstract
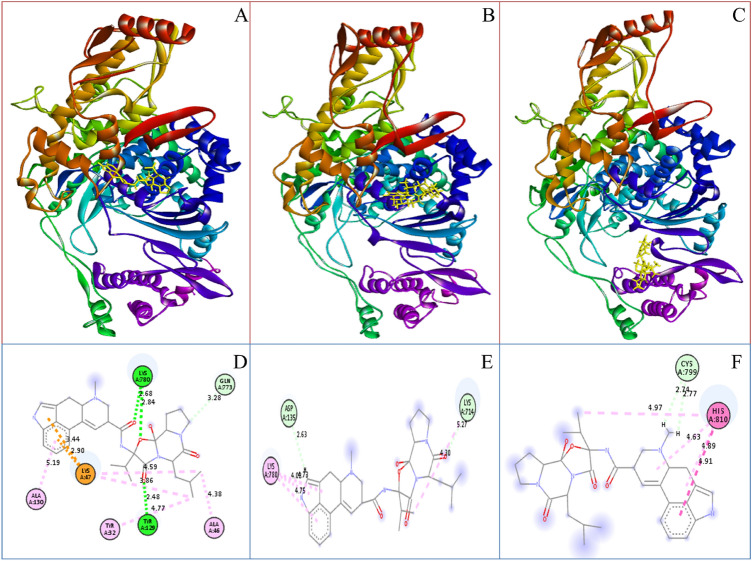
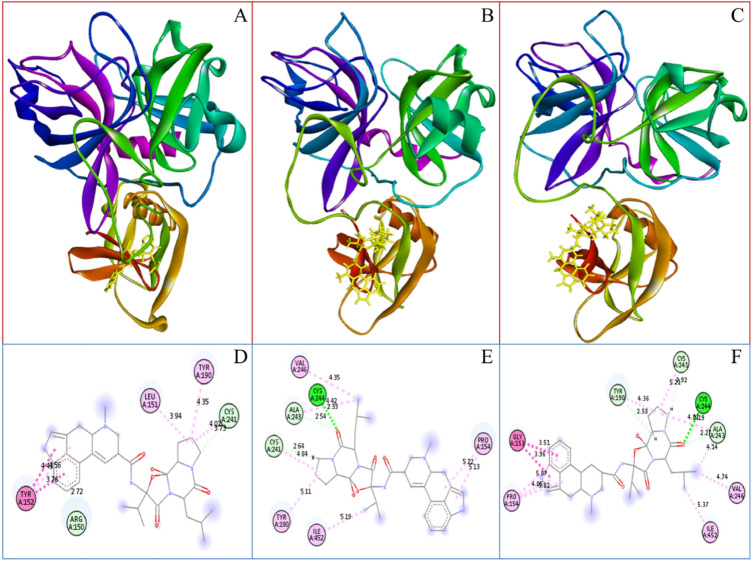
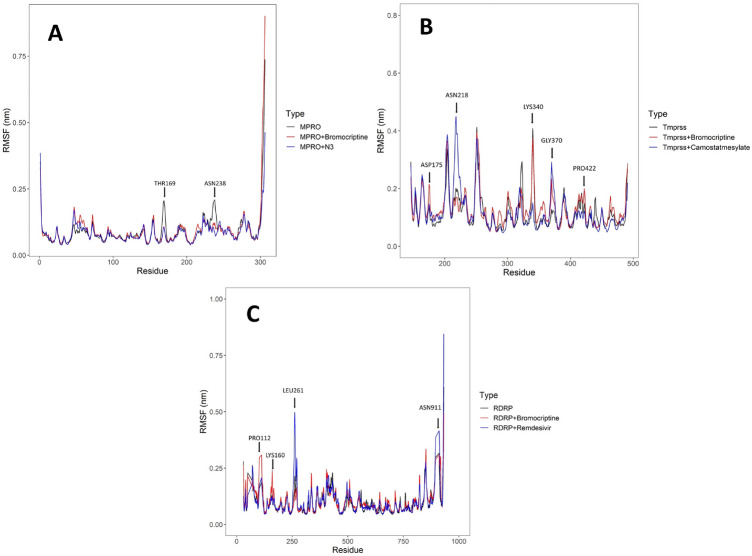
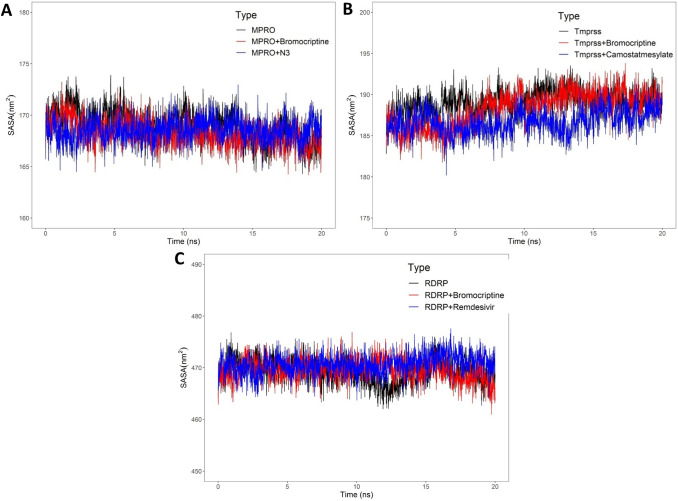
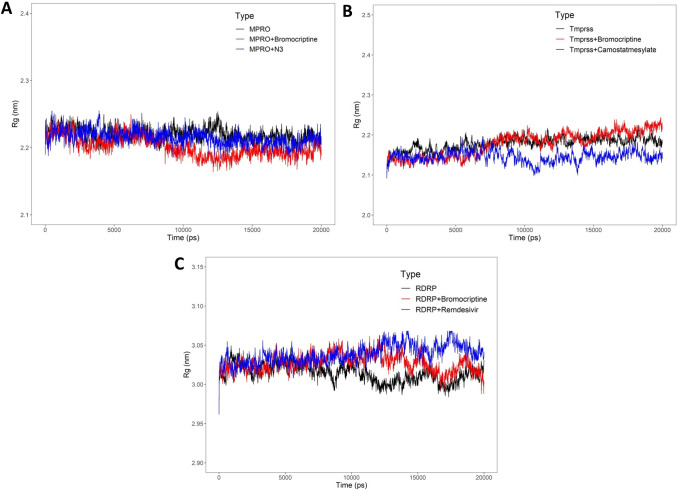
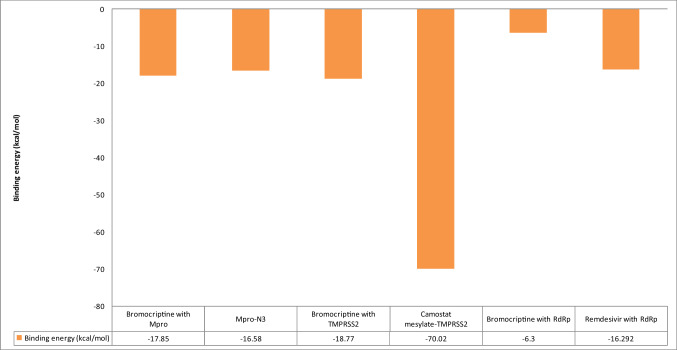
Coronavirus spread is an emergency reported globally, and a specific treatment strategy for this significant health issue is not yet identified. COVID-19 is a highly contagious disease and needs to be controlled promptly as millions of deaths have been reported. Due to the absence of proficient restorative alternatives and preliminary clinical restrictions, FDA-approved medications can be a decent alternative to deal with the coronavirus malady (COVID-19). The present study aims to meet the imperative necessity of effective COVID-19 drug treatment with a computational multi-target drug repurposing approach. This study focused on screening the FDA-approved drugs derived from the fungal source and its derivatives against the SARS-CoV-2 targets. All the selected drugs showed good binding affinity towards these targets, and out of them, bromocriptine was found to be the best candidate after the screening on the COVID-19 targets. Further, bromocriptine is analyzed by molecular simulation and MM-PBSA study. These studies suggested that bromocriptine can be the best candidate for TMPRSS2, Main protease, and RdRp protein.
Supplementary information: The online version contains supplementary material available at 10.1007/s40203-021-00089-8.
Keywords: And molecular simulation; COVID-19; Drug repurposing; FDA approved drugs; Molecular docking.
© The Author(s), under exclusive licence to Springer-Verlag GmbH Germany, part of Springer Nature 2021.
Figures

Similar articles
-
In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing.J Infect Public Health. 2020 Sep;13(9):1210-1223. doi: 10.1016/j.jiph.2020.06.016. Epub 2020 Jun 16. J Infect Public Health. 2020. PMID: 32561274 Free PMC article.
-
Computational screening of dual inhibitors from FDA approved antiviral drugs on SARS-CoV-2 spike protein and the main protease using molecular docking approach.Acta Virol. 2021;65(2):160-172. doi: 10.4149/av_2021_208. Acta Virol. 2021. PMID: 34130467
-
Potential protease inhibitors and their combinations to block SARS-CoV-2.J Biomol Struct Dyn. 2022 Feb;40(2):903-917. doi: 10.1080/07391102.2020.1819881. Epub 2020 Sep 14. J Biomol Struct Dyn. 2022. PMID: 32924827 Free PMC article.
-
Computational insights into binding mechanism of drugs as potential inhibitors against SARS-CoV-2 targets.Chem Zvesti. 2022;76(1):111-121. doi: 10.1007/s11696-021-01843-0. Epub 2021 Aug 30. Chem Zvesti. 2022. PMID: 34483461 Free PMC article.
-
Hybrid drug-screening strategy identifies potential SARS-CoV-2 cell-entry inhibitors targeting human transmembrane serine protease.Struct Chem. 2022;33(5):1503-1515. doi: 10.1007/s11224-022-01960-w. Epub 2022 May 11. Struct Chem. 2022. PMID: 35571866 Free PMC article.
Cited by
-
Binary-QSAR guided virtual screening of FDA approved drugs and compounds in clinical investigation against SARS-CoV-2 main protease.Turk J Biol. 2021 Aug 30;45(4):459-468. doi: 10.3906/biy-2106-61. eCollection 2021. Turk J Biol. 2021. PMID: 34803447 Free PMC article.
-
Screening for inhibitors against SARS-CoV-2 and its variants.Biosaf Health. 2022 Jun;4(3):186-192. doi: 10.1016/j.bsheal.2022.05.002. Epub 2022 May 7. Biosaf Health. 2022. PMID: 35574239 Free PMC article.
-
An insight into SARS-CoV-2 structure, pathogenesis, target hunting for drug development and vaccine initiatives.RSC Med Chem. 2022 Jan 25;13(6):647-675. doi: 10.1039/d2md00009a. eCollection 2022 Jun 22. RSC Med Chem. 2022. PMID: 35814927 Free PMC article. Review.
-
Network analysis-guided drug repurposing strategies targeting LPAR receptor in the interplay of COVID, Alzheimer's, and diabetes.Sci Rep. 2024 Feb 21;14(1):4328. doi: 10.1038/s41598-024-55013-9. Sci Rep. 2024. PMID: 38383841 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
