

miR-181d/RBP2/NF-κB p65 Feedback Regulation Promotes Chronic Myeloid Leukemia Blast Crisis
- PMID: 33842368
- PMCID: PMC8027495
- DOI: 10.3389/fonc.2021.654411
miR-181d/RBP2/NF-κB p65 Feedback Regulation Promotes Chronic Myeloid Leukemia Blast Crisis
Abstract
Background: Chronic myeloid leukemia (CML) is a malignant clonal proliferative disease. Once it progresses into the phase of blast crisis (CML-BP), the curative effect is poor, and the fatality rate is extremely high. Therefore, it is urgent to explore the molecular mechanisms of blast crisis and identify new therapeutic targets.
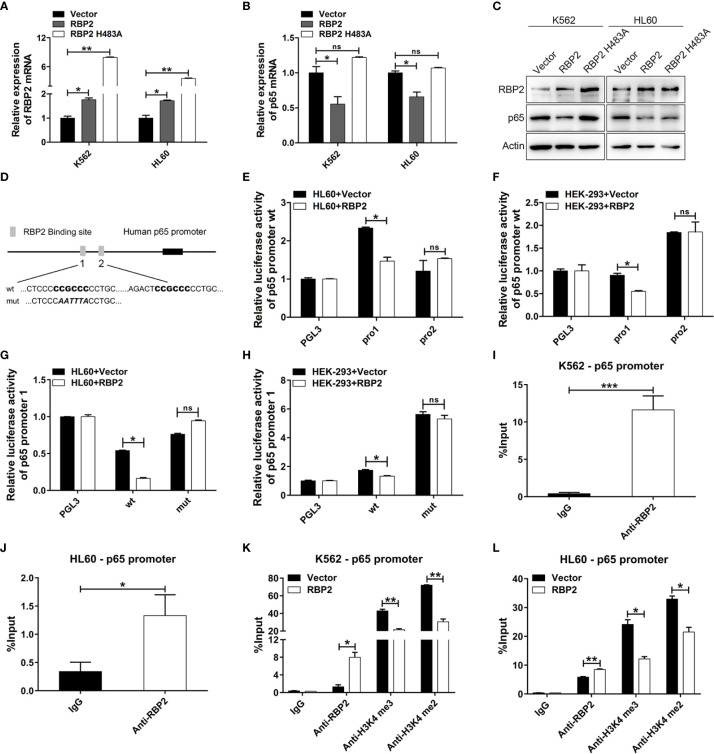
Methods: The expression levels of miR-181d, RBP2 and NF-κB p65 were assessed in 42 newly diagnosed CML-CP patients and 15 CML-BP patients. Quantitative real-time PCR, Western blots, and cell proliferation assay were used to characterize the changes induced by overexpression or inhibition of miR-181d, RBP2 or p65. Luciferase reporter assay and ChIP assay was conducted to establish functional association between miR-181d, RBP2 and p65. Inhibition of miR-181d expression and its consequences in tumor growth was demonstrated in vivo models.
Results: We found that miR-181d was overexpressed in CML-BP, which promoted leukemia cell proliferation. Histone demethylase RBP2 was identified as a direct target of miR-181d which downregulated RBP2 expression. Moreover, RBP2 inhibited transcriptional expression of NF-κB subunit, p65 by binding to its promoter and demethylating the tri/dimethylated H3K4 region in the p65 promoter locus. In turn, p65 directly bound to miR-181d promoter and upregulated its expression. Therefore, RBP2 inhibition resulting from miR-181d overexpression led to p65 upregulation which further forwarded miR-181d expression. This miR-181d/RBP2/p65 feedback regulation caused sustained NF-κB activation, which contributed to the development of CML-BP.
Conclusions: Taken together, the miR-181d/RBP2/p65 feedback regulation promoted CML-BP and miR-181d may serve as a potential therapeutic target of CML-BP.
Keywords: CML blast crisis; RBP2; cell proliferation; miR-181d; p65.
Copyright © 2021 Zhou, Yin, Zheng, Fu, Wang, Cui, Gao, Wang, Huang, Jia and Chen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





Similar articles
-
Histone demethylase RBP2 decreases miR-21 in blast crisis of chronic myeloid leukemia.Oncotarget. 2015 Jan 20;6(2):1249-61. doi: 10.18632/oncotarget.2859. Oncotarget. 2015. PMID: 25575817 Free PMC article.
-
RNA-Binding Protein Lin28B Promotes Chronic Myeloid Leukemia Blast Crisis by Transcriptionally Upregulating miR-181d.Mol Cancer Res. 2024 Oct 2;22(10):932-942. doi: 10.1158/1541-7786.MCR-23-0928. Mol Cancer Res. 2024. PMID: 38847604
-
Histone demethylase RBP2 mediates the blast crisis of chronic myeloid leukemia through an RBP2/PTEN/BCR-ABL cascade.Cell Signal. 2019 Nov;63:109360. doi: 10.1016/j.cellsig.2019.109360. Epub 2019 Jul 30. Cell Signal. 2019. PMID: 31374292
-
miR-181d regulates human dendritic cell maturation through NF-κB pathway.Cell Prolif. 2017 Oct;50(5):e12358. doi: 10.1111/cpr.12358. Epub 2017 Jul 21. Cell Prolif. 2017. PMID: 28731516 Free PMC article.
-
Histone demethylase retinoblastoma binding protein 2 is overexpressed in hepatocellular carcinoma and negatively regulated by hsa-miR-212.PLoS One. 2013 Jul 29;8(7):e69784. doi: 10.1371/journal.pone.0069784. Print 2013. PLoS One. 2013. PMID: 23922798 Free PMC article.
Cited by
-
Role of miRNAs to control the progression of Chronic Myeloid Leukemia by their expression levels.Med Oncol. 2024 Jan 12;41(2):55. doi: 10.1007/s12032-023-02278-1. Med Oncol. 2024. PMID: 38216843 Review.
-
Bioinformatic Tools for the Analysis and Prediction of ncRNA Interactions.Int J Mol Sci. 2021 Oct 22;22(21):11397. doi: 10.3390/ijms222111397. Int J Mol Sci. 2021. PMID: 34768830 Free PMC article. Review.
-
Epitranscriptomics in the development, functions, and disorders of cancer stem cells.Front Oncol. 2023 Mar 17;13:1145766. doi: 10.3389/fonc.2023.1145766. eCollection 2023. Front Oncol. 2023. PMID: 37007137 Free PMC article. Review.
-
DNA methylation-mediated differential expression of DLX4 isoforms has opposing roles in leukemogenesis.Cell Mol Biol Lett. 2022 Jul 26;27(1):59. doi: 10.1186/s11658-022-00358-0. Cell Mol Biol Lett. 2022. PMID: 35883028 Free PMC article.
-
Role of main RNA modifications in cancer: N6-methyladenosine, 5-methylcytosine, and pseudouridine.Signal Transduct Target Ther. 2022 Apr 28;7(1):142. doi: 10.1038/s41392-022-01003-0. Signal Transduct Target Ther. 2022. PMID: 35484099 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous