

Soft Materials by Design: Unconventional Polymer Networks Give Extreme Properties
- PMID: 33844906
- PMCID: PMC9217625
- DOI: 10.1021/acs.chemrev.0c01088
Soft Materials by Design: Unconventional Polymer Networks Give Extreme Properties
Abstract
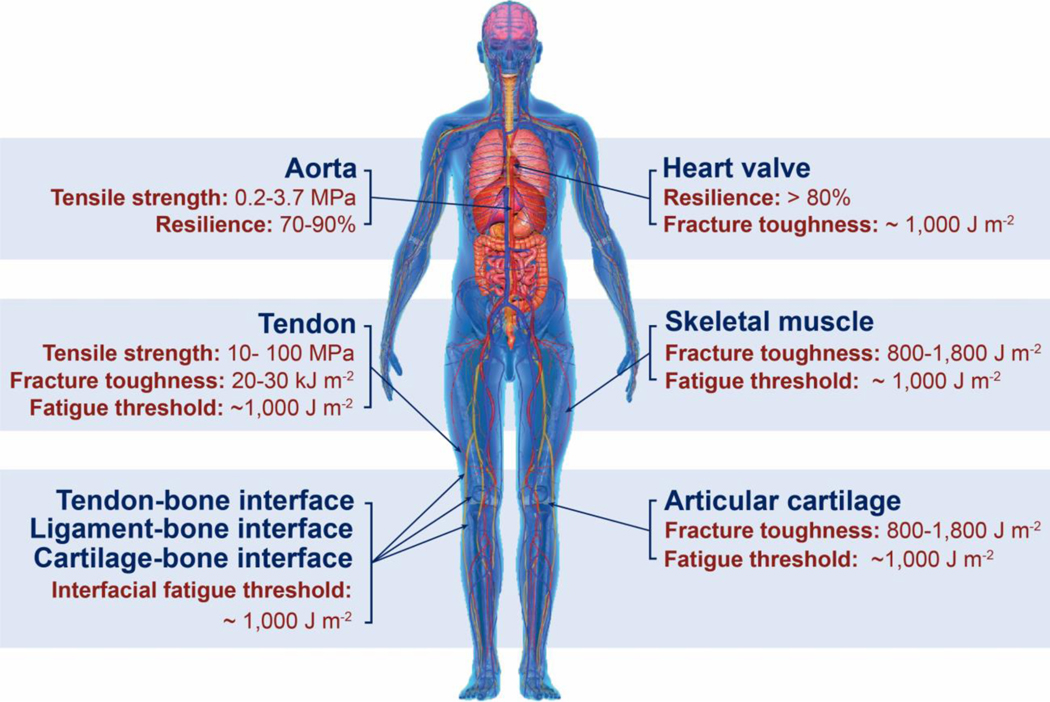
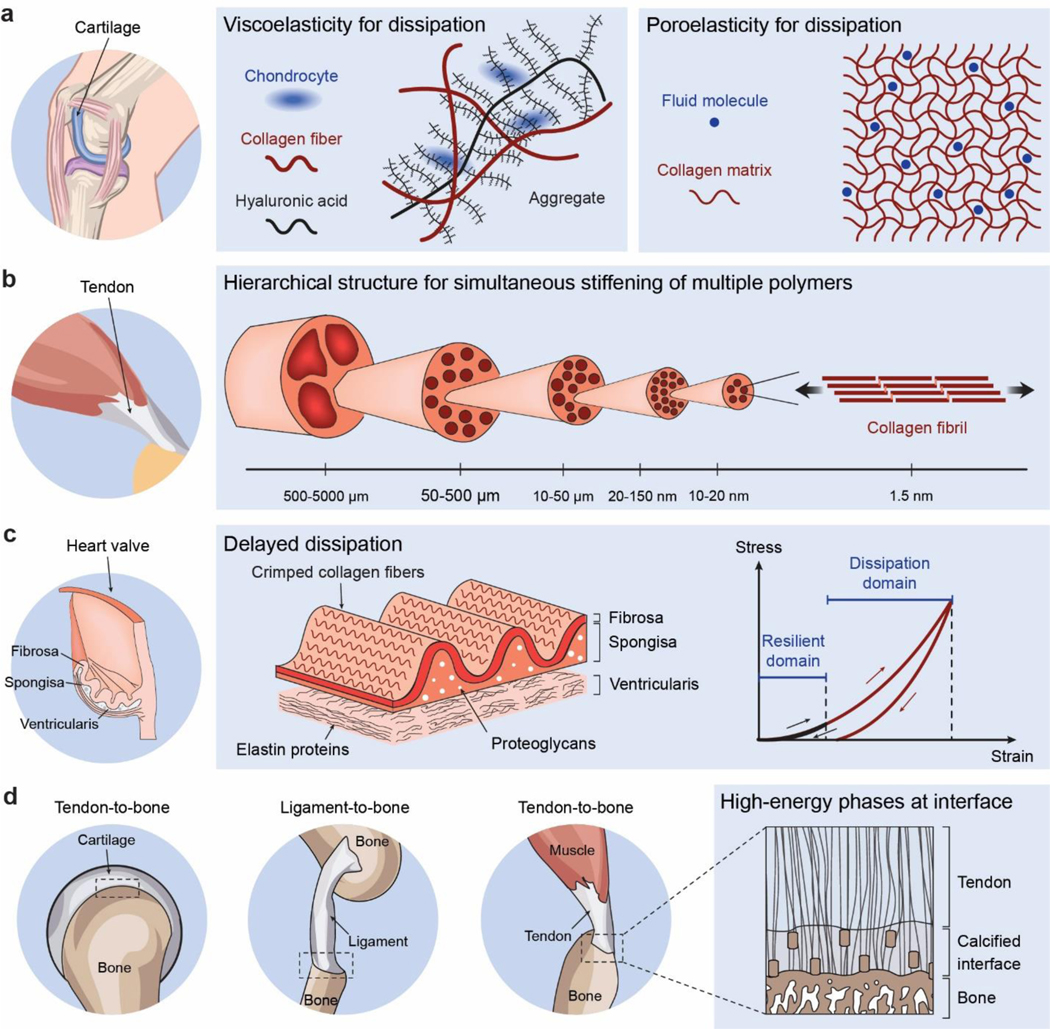
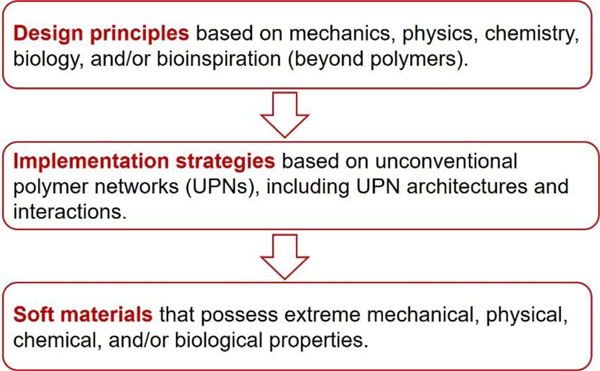
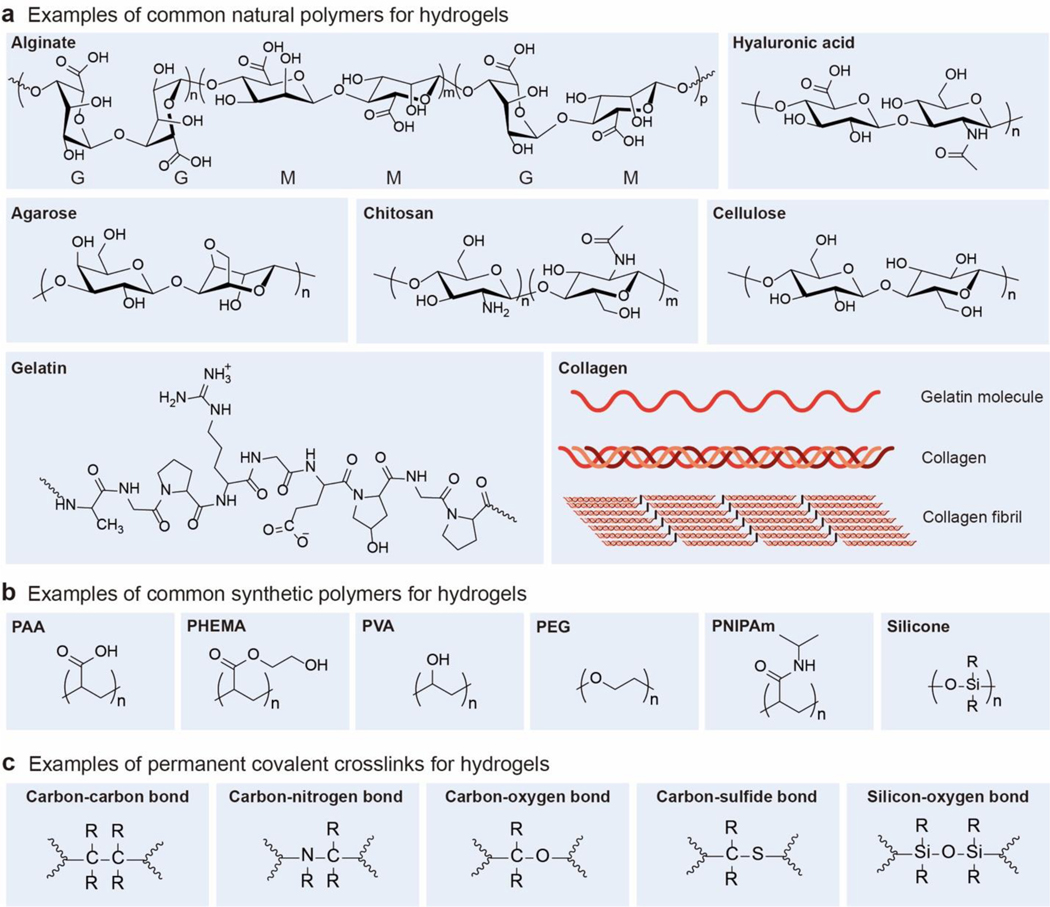
Hydrogels are polymer networks infiltrated with water. Many biological hydrogels in animal bodies such as muscles, heart valves, cartilages, and tendons possess extreme mechanical properties including being extremely tough, strong, resilient, adhesive, and fatigue-resistant. These mechanical properties are also critical for hydrogels' diverse applications ranging from drug delivery, tissue engineering, medical implants, wound dressings, and contact lenses to sensors, actuators, electronic devices, optical devices, batteries, water harvesters, and soft robots. Whereas numerous hydrogels have been developed over the last few decades, a set of general principles that can rationally guide the design of hydrogels using different materials and fabrication methods for various applications remain a central need in the field of soft materials. This review is aimed at synergistically reporting: (i) general design principles for hydrogels to achieve extreme mechanical and physical properties, (ii) implementation strategies for the design principles using unconventional polymer networks, and (iii) future directions for the orthogonal design of hydrogels to achieve multiple combined mechanical, physical, chemical, and biological properties. Because these design principles and implementation strategies are based on generic polymer networks, they are also applicable to other soft materials including elastomers and organogels. Overall, the review will not only provide comprehensive and systematic guidelines on the rational design of soft materials, but also provoke interdisciplinary discussions on a fundamental question: why does nature select soft materials with unconventional polymer networks to constitute the major parts of animal bodies?
Figures
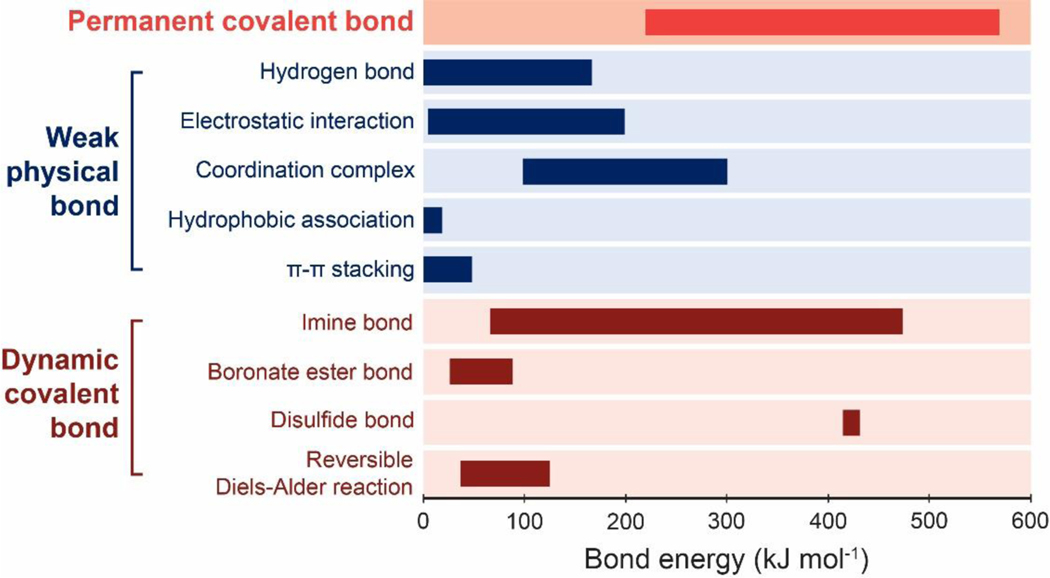
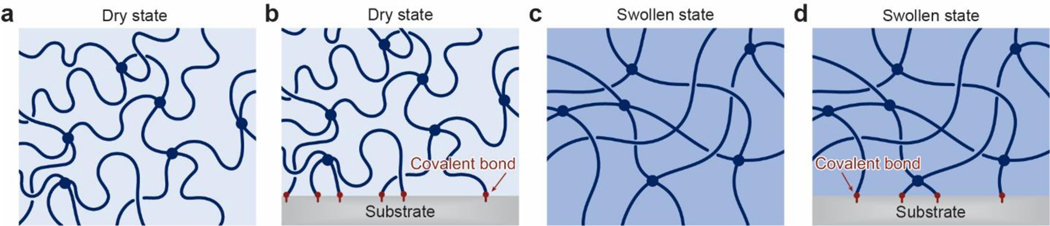
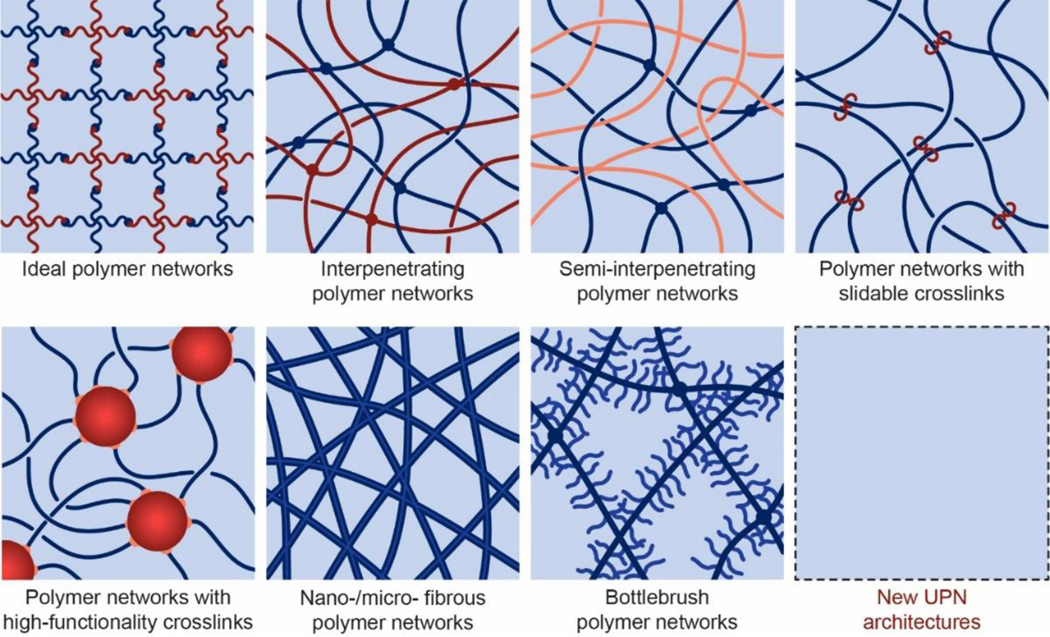
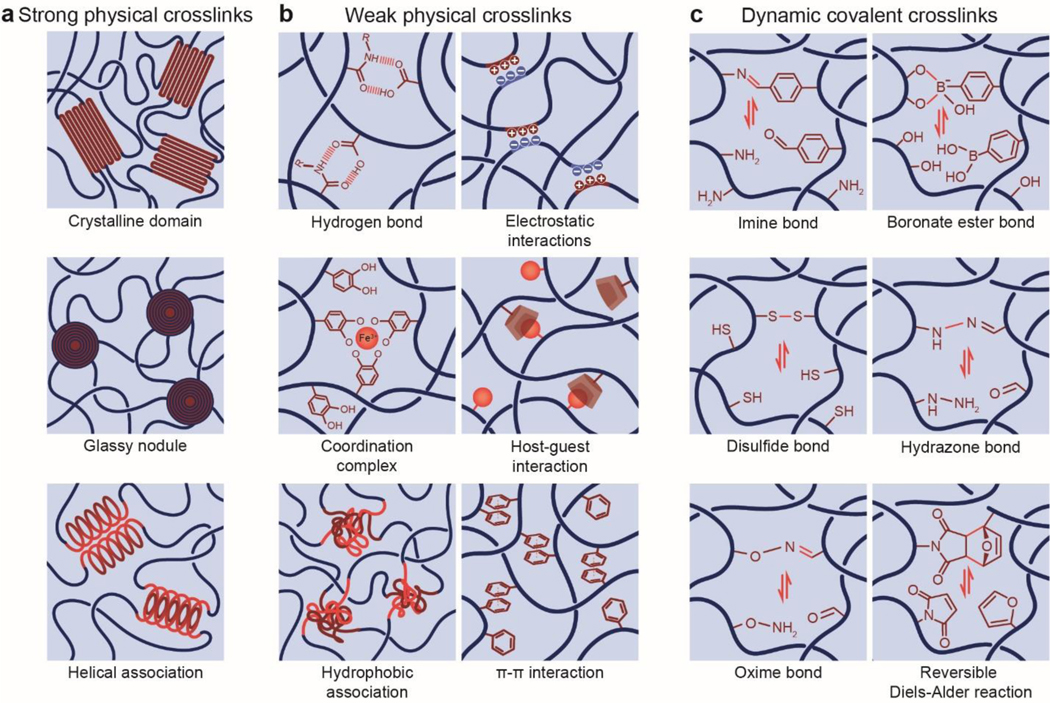
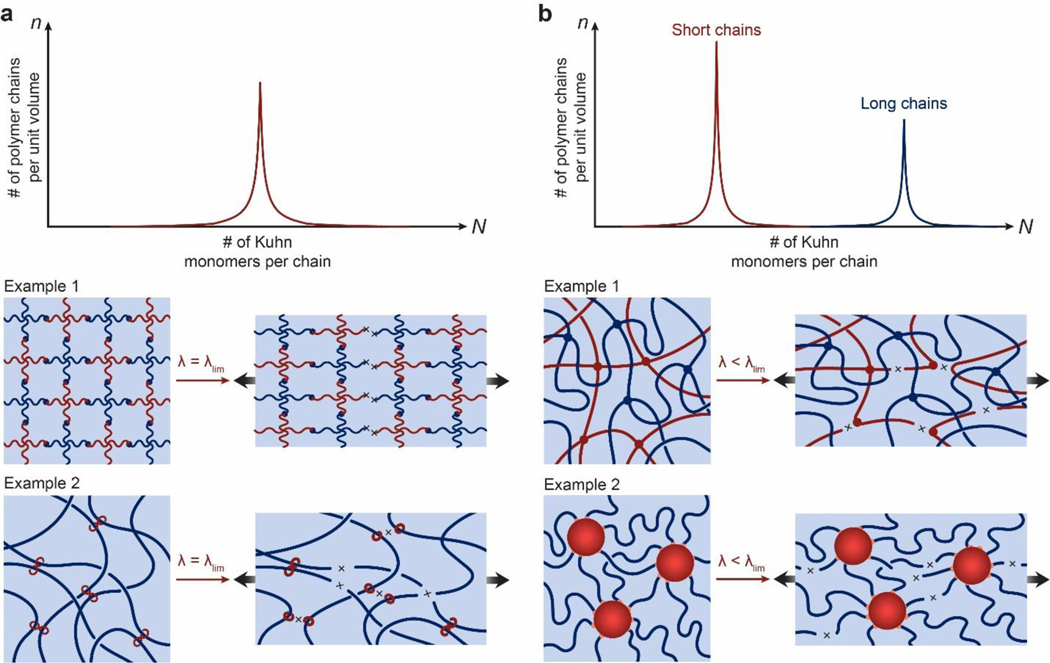
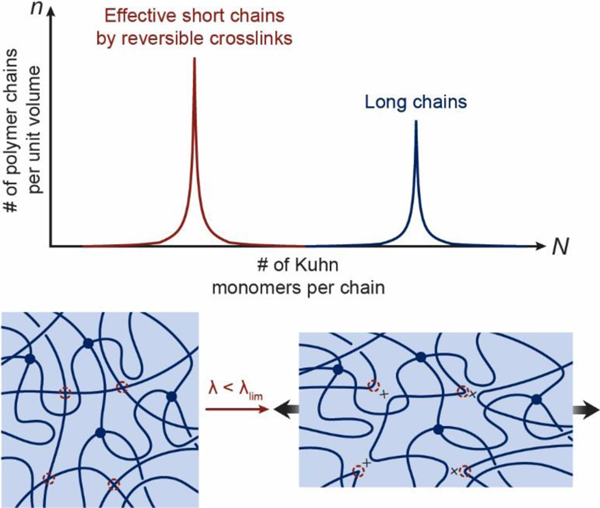
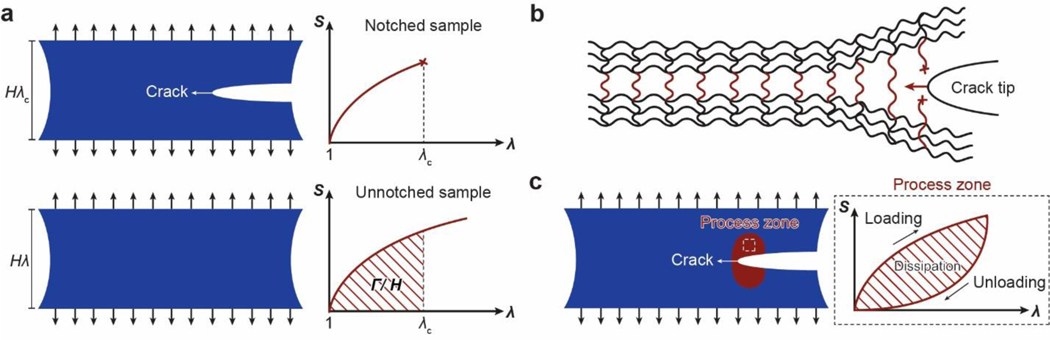
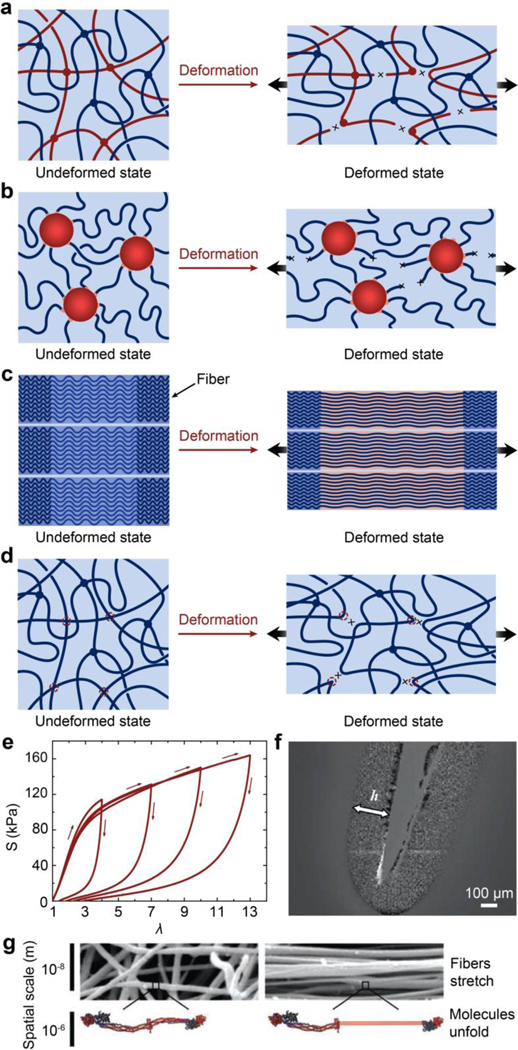
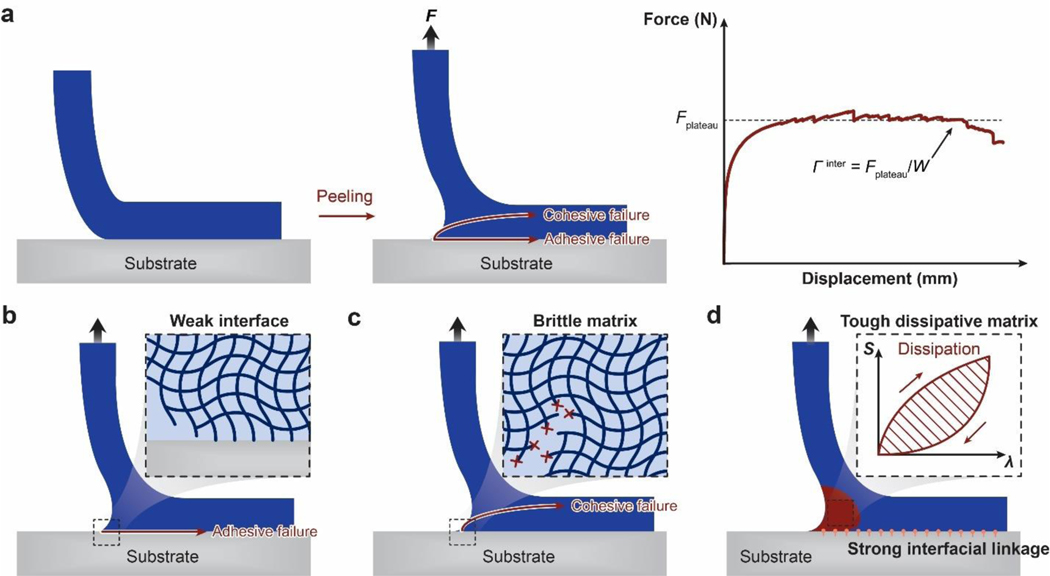
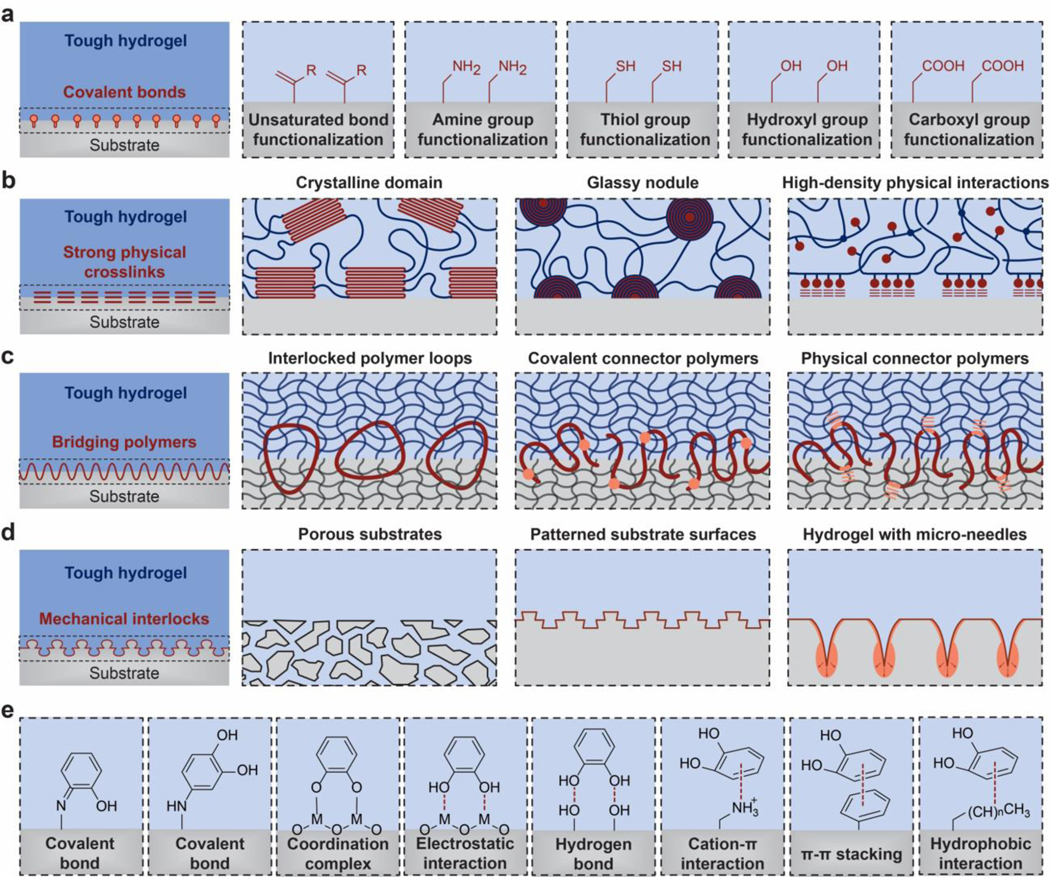
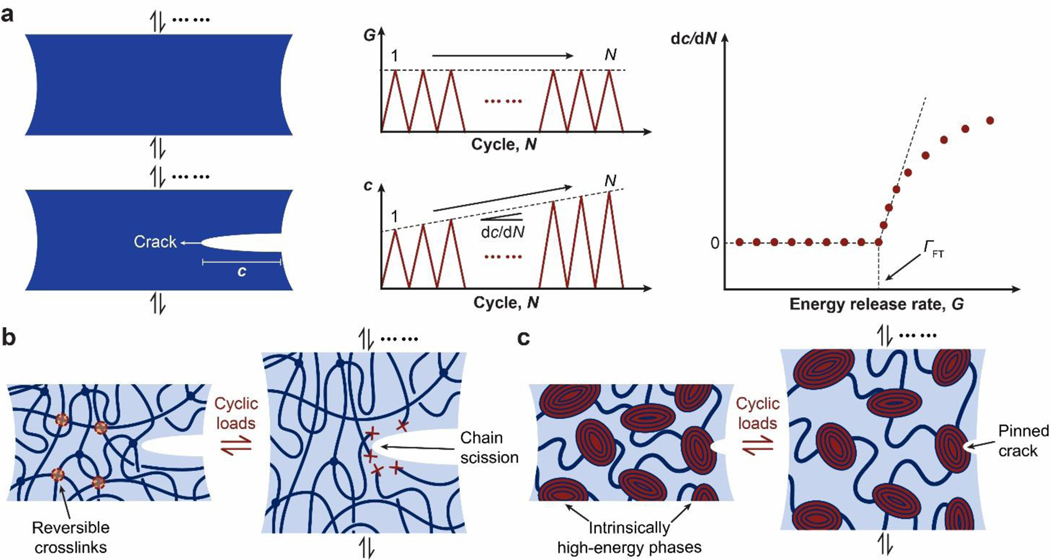
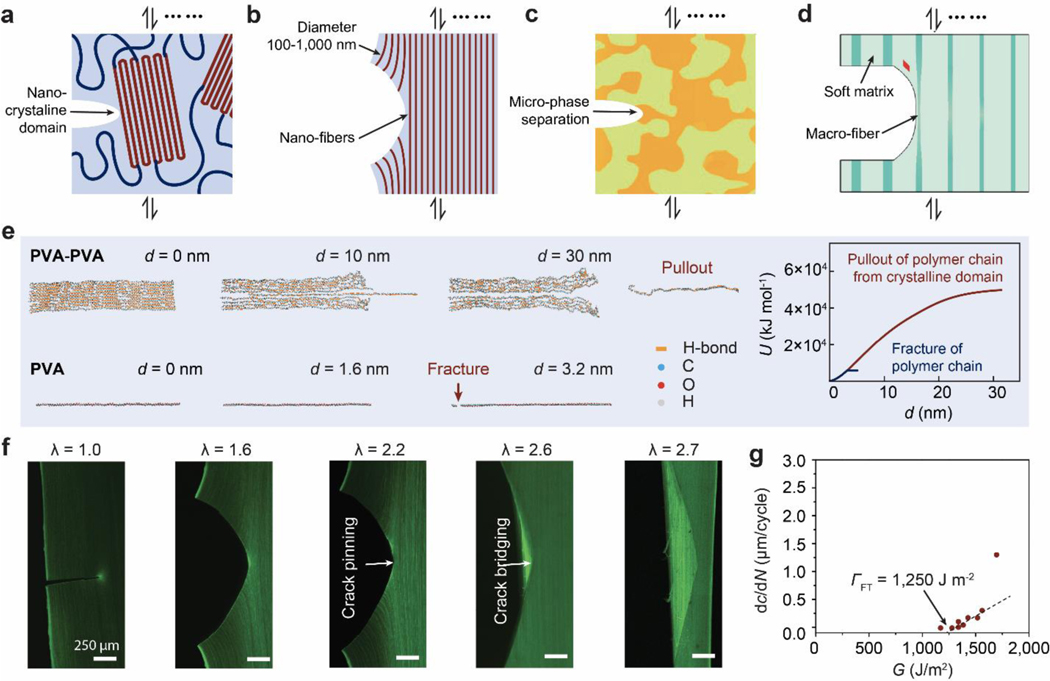
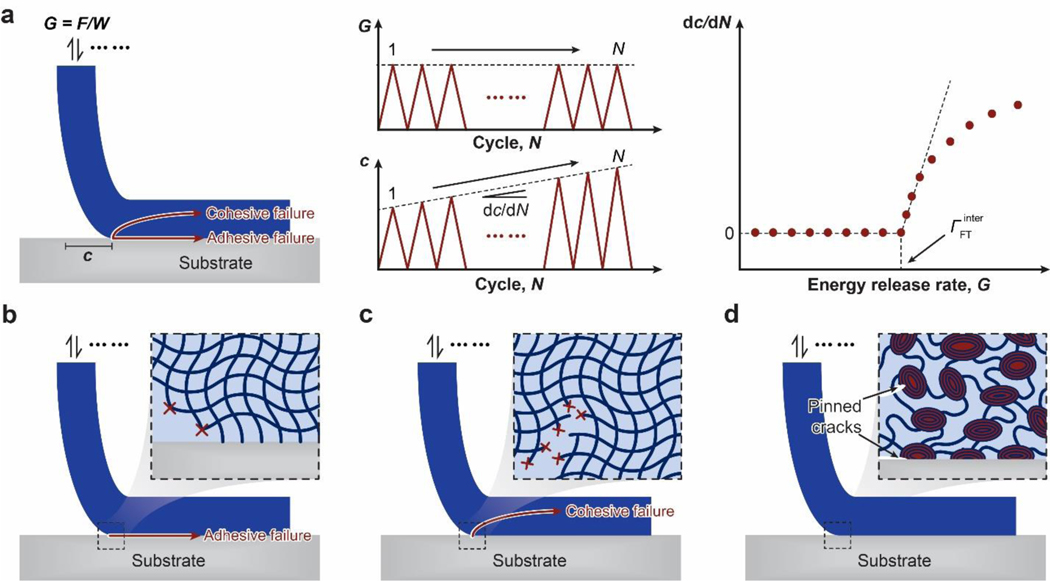
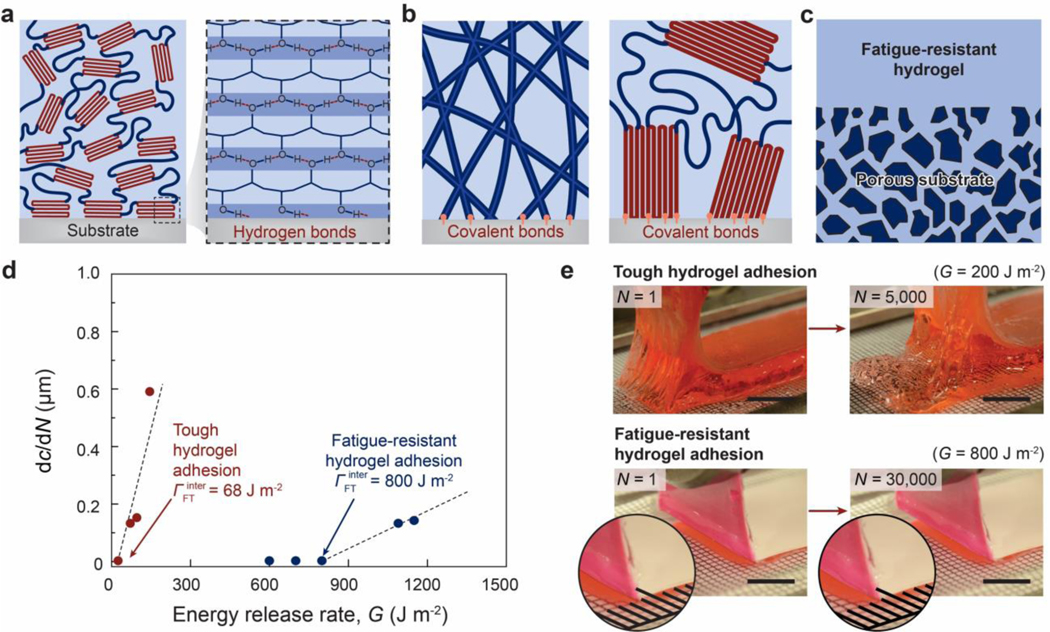
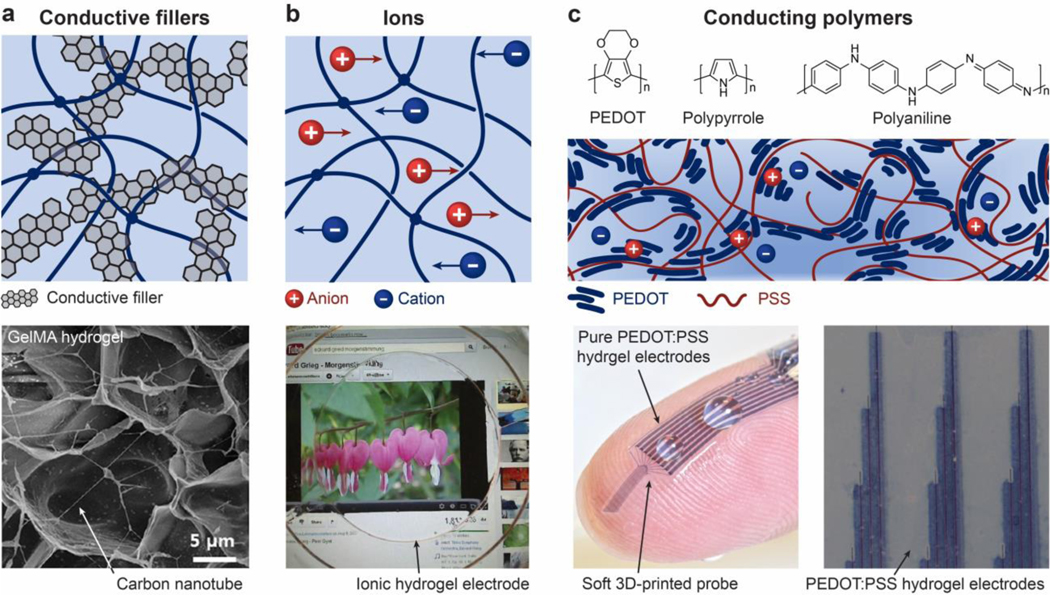
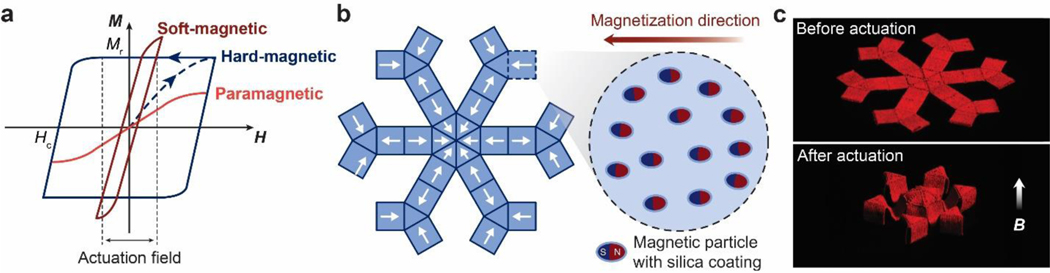
References
-
- Wichterle O; LÍM D.Hydrophilic Gels for Biological Use. Nature 1960, 185 (4706), 117.
-
- Peppas NA; Bures P; Leobandung W; Ichikawa H.Hydrogels in pharmaceutical formulations. European Journal of Pharmaceutics and Biopharmaceutics 2000, 50 (1), 27. - PubMed
-
- Qiu Y; Park K.Environment-sensitive hydrogels for drug delivery. Advanced Drug Delivery Reviews 2001, 53 (3), 321. - PubMed
-
- Peppas NA; Hilt JZ; Khademhosseini A; Langer R.Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Advanced Materials 2006, 18 (11), 1345.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
