

Isolation and characterisation of ΦcrAss002, a crAss-like phage from the human gut that infects Bacteroides xylanisolvens
- PMID: 33845877
- PMCID: PMC8042965
- DOI: 10.1186/s40168-021-01036-7
Isolation and characterisation of ΦcrAss002, a crAss-like phage from the human gut that infects Bacteroides xylanisolvens
Abstract
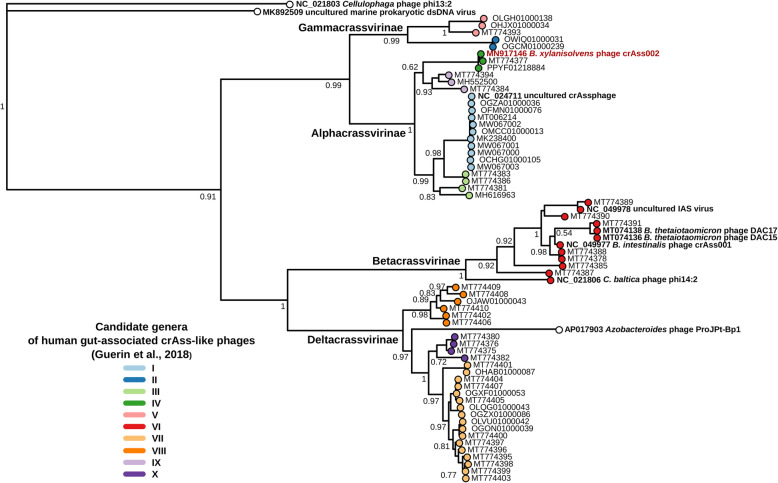
Background: The gut phageome comprises a complex phage community of thousands of individual strains, with a few highly abundant bacteriophages. CrAss-like phages, which infect bacteria of the order Bacteroidales, are the most abundant bacteriophage family in the human gut and make an important contribution to an individual's core virome. Based on metagenomic data, crAss-like phages form a family, with four sub-families and ten candidate genera. To date, only three representatives isolated in pure culture have been reported: ΦcrAss001 and two closely related phages DAC15 and DAC17; all are members of the less abundant candidate genus VI. The persistence at high levels of both crAss-like phage and their Bacteroidales hosts in the human gut has not been explained mechanistically, and this phage-host relationship can only be properly studied with isolated phage-host pairs from as many genera as possible.
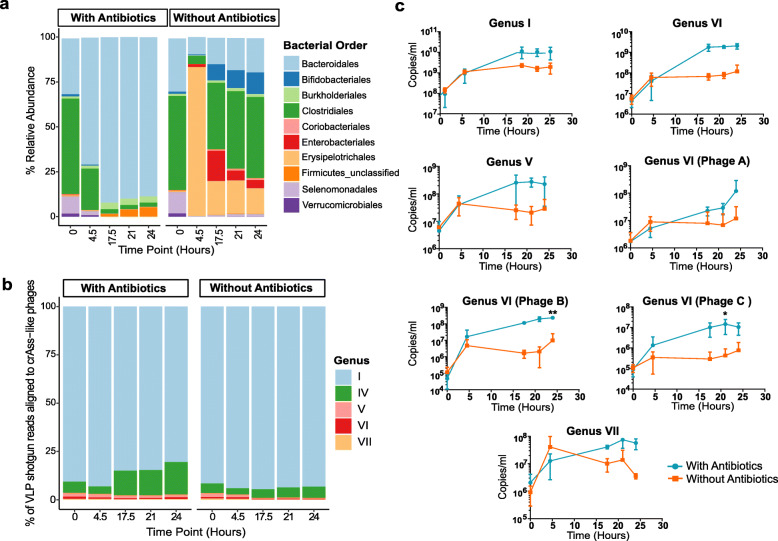
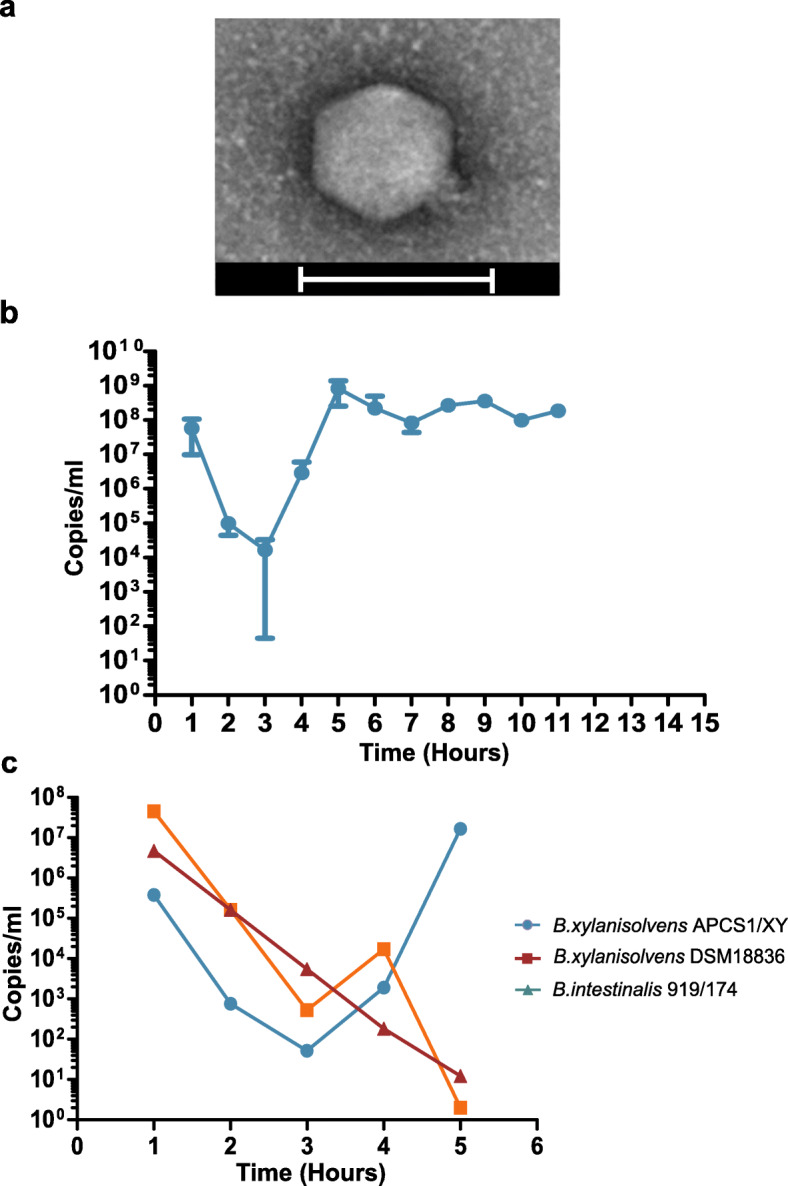
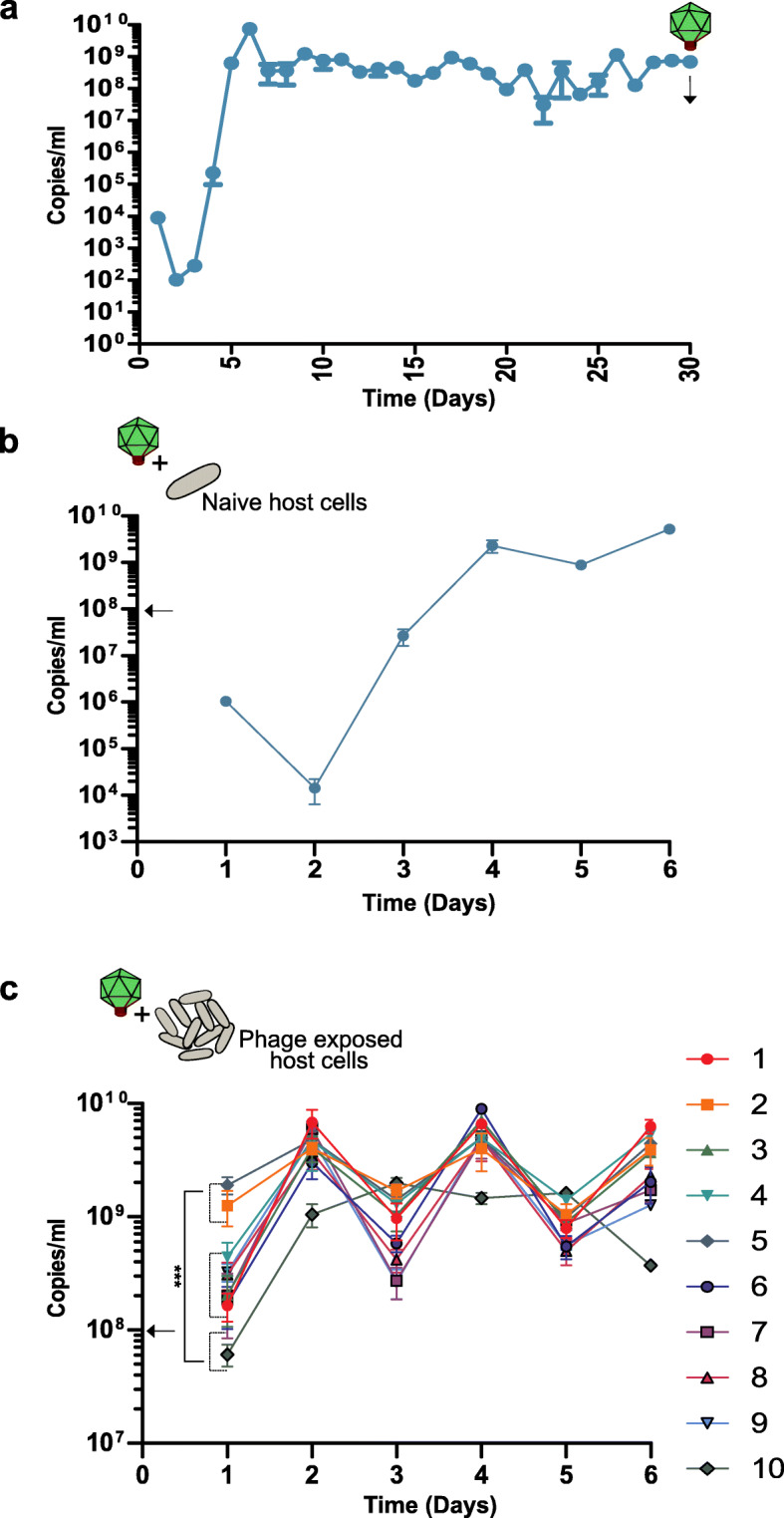
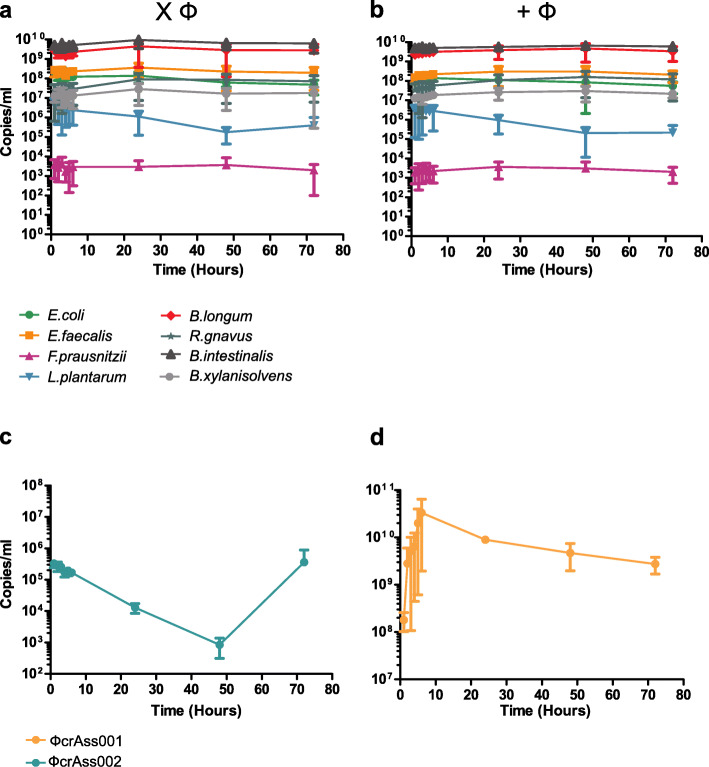
Results: Faeces from a healthy donor with high levels of crAss-like phage was used to initiate a faecal fermentation in a chemostat, with selected antibiotics chosen to inhibit rapidly growing bacteria and selectively enrich for Gram-negative Bacteroidales. This had the objective of promoting the simultaneous expansion of crAss-like phages on their native hosts. The levels of seven different crAss-like phages expanded during the fermentation, indicating that their hosts were also present in the fermenter. The enriched supernatant was then tested against individual Bacteroidales strains isolated from the same faecal sample. This resulted in the isolation of a previously uncharacterised crAss-like phage of candidate genus IV of the proposed Alphacrassvirinae sub-family, ΦcrAss002, that infects the gut commensal Bacteroides xylanisolvens. ΦcrAss002 does not form plaques or spots on lawns of sensitive cells, nor does it lyse liquid cultures, even at high titres. In keeping with the co-abundance of phage and host in the human gut, ΦcrAss002 and Bacteroides xylanisolvens can also co-exist at high levels when co-cultured in laboratory media.
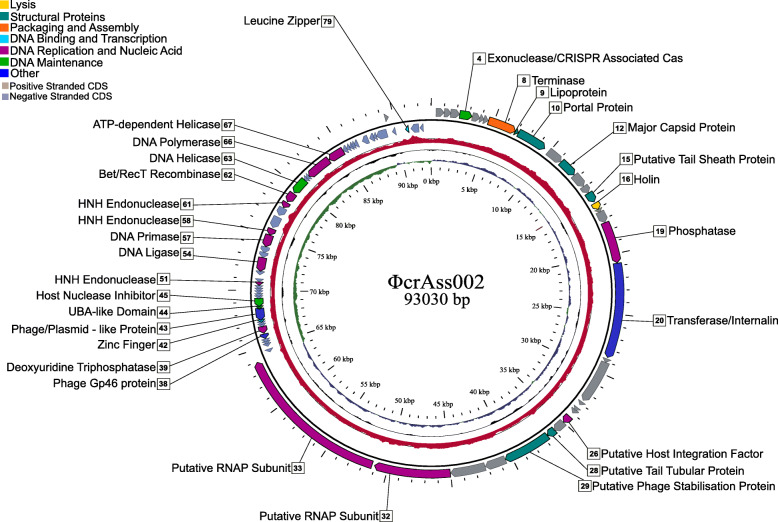
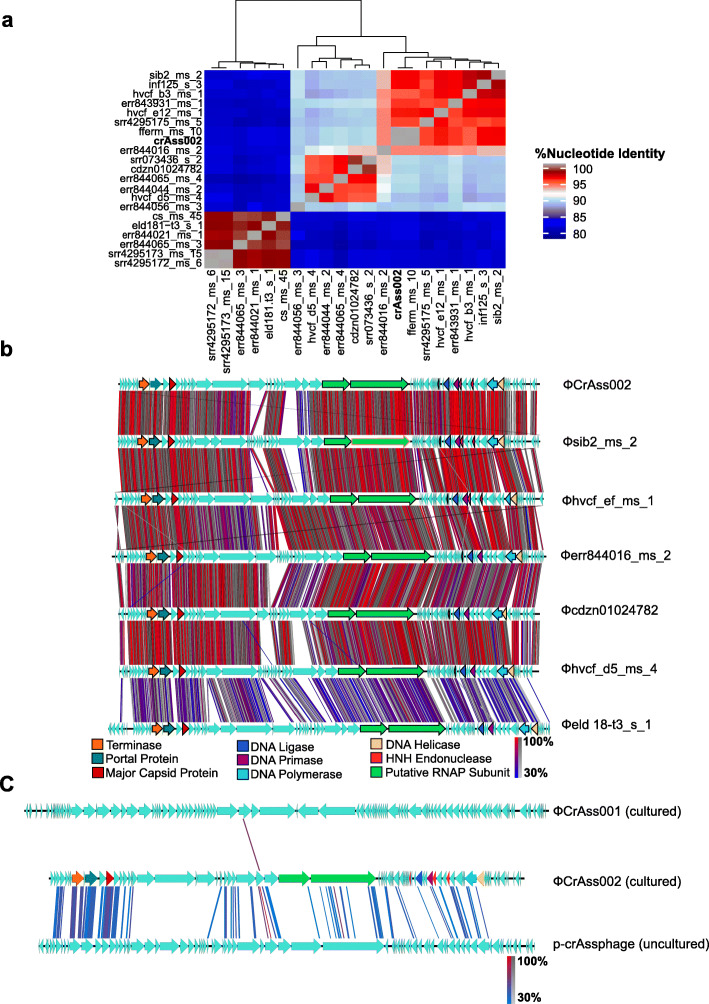
Conclusions: We report the isolation and characterisation of ΦcrAss002, the first representative of the proposed Alphacrassvirinae sub-family of crAss-like phages. ΦcrAss002 cannot form plaques or spots on bacterial lawns but can co-exist with its host, Bacteroides xylanisolvens, at very high levels in liquid culture without impacting on bacterial numbers. Video abstract.
Keywords: Bacteriophages; Human gut phageome; Human microbiome; Phage-host interactions; crAss-like phages; crAssphage.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
