

Learning cis-regulatory principles of ADAR-based RNA editing from CRISPR-mediated mutagenesis
- PMID: 33846332
- PMCID: PMC8041805
- DOI: 10.1038/s41467-021-22489-2
Learning cis-regulatory principles of ADAR-based RNA editing from CRISPR-mediated mutagenesis
Abstract
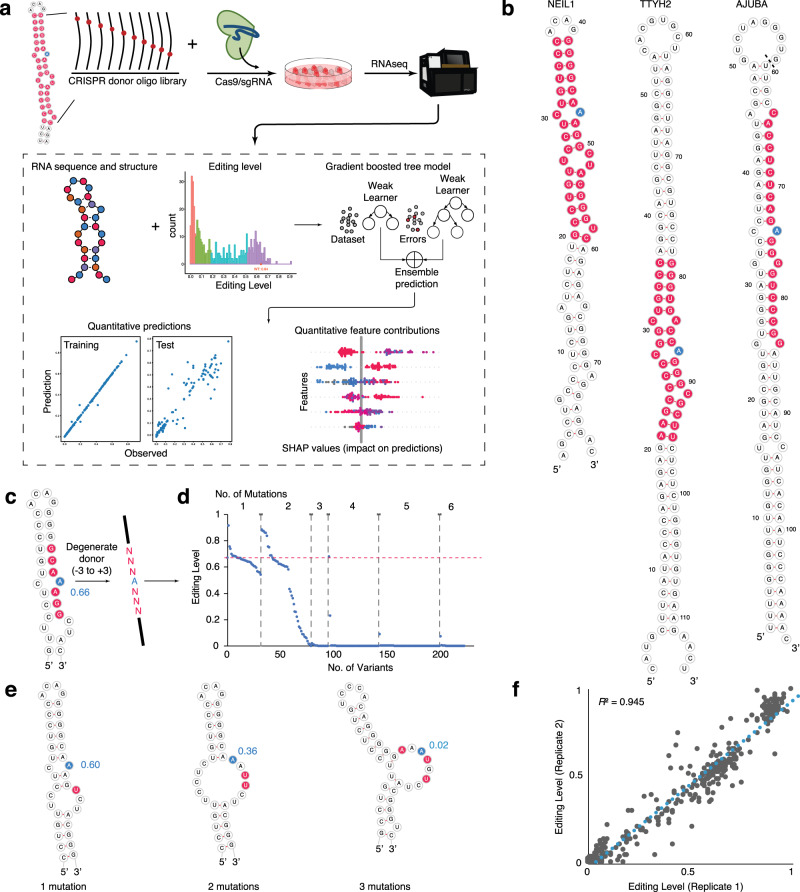
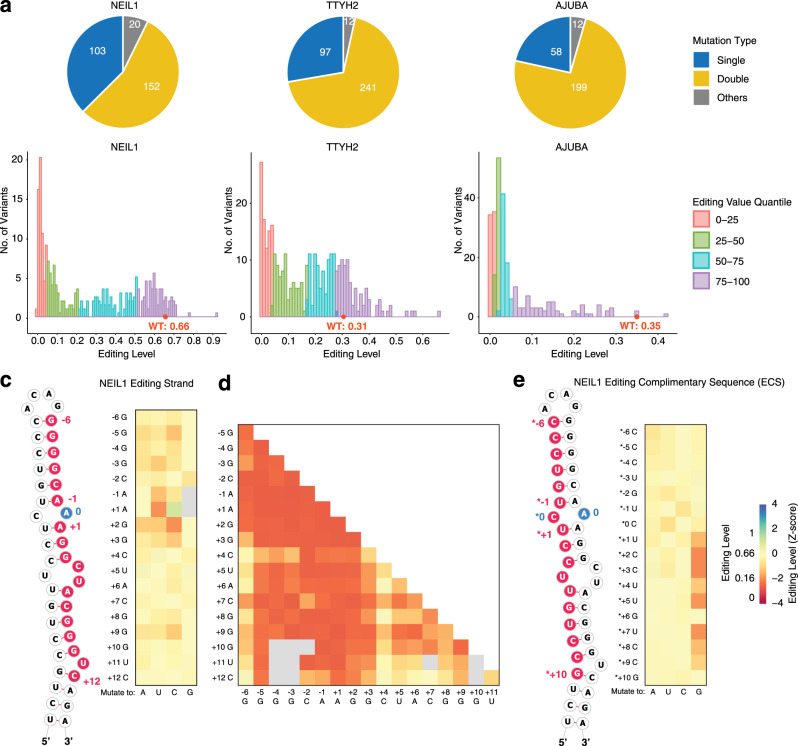
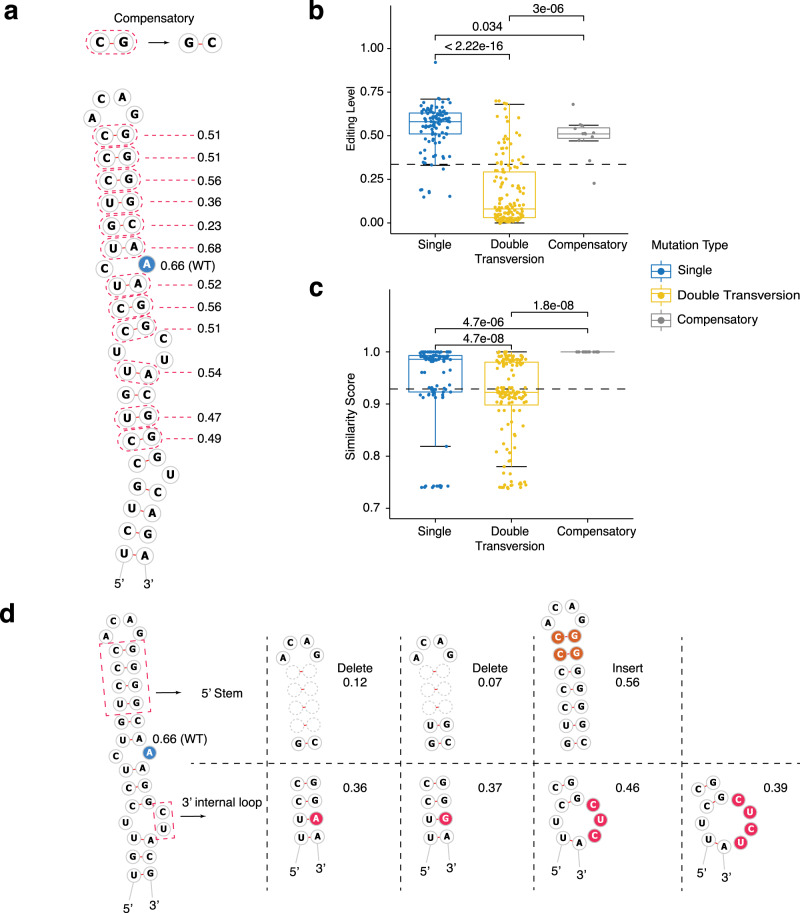
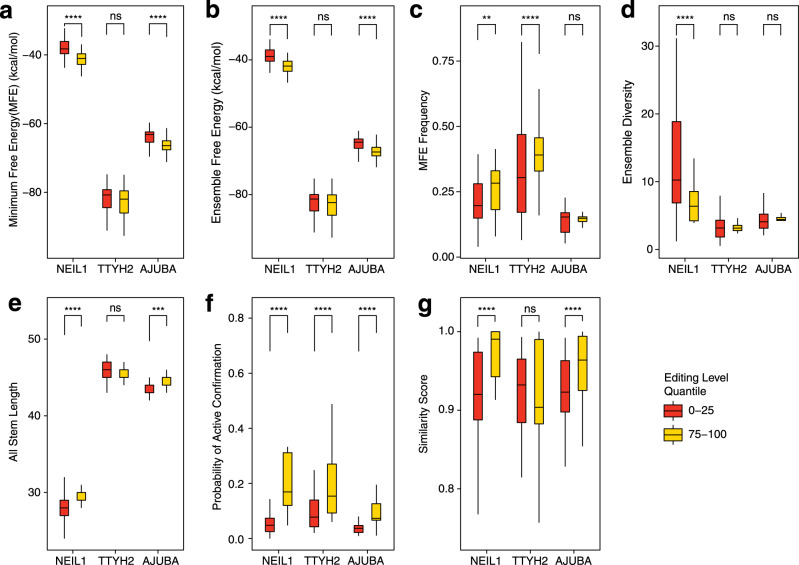
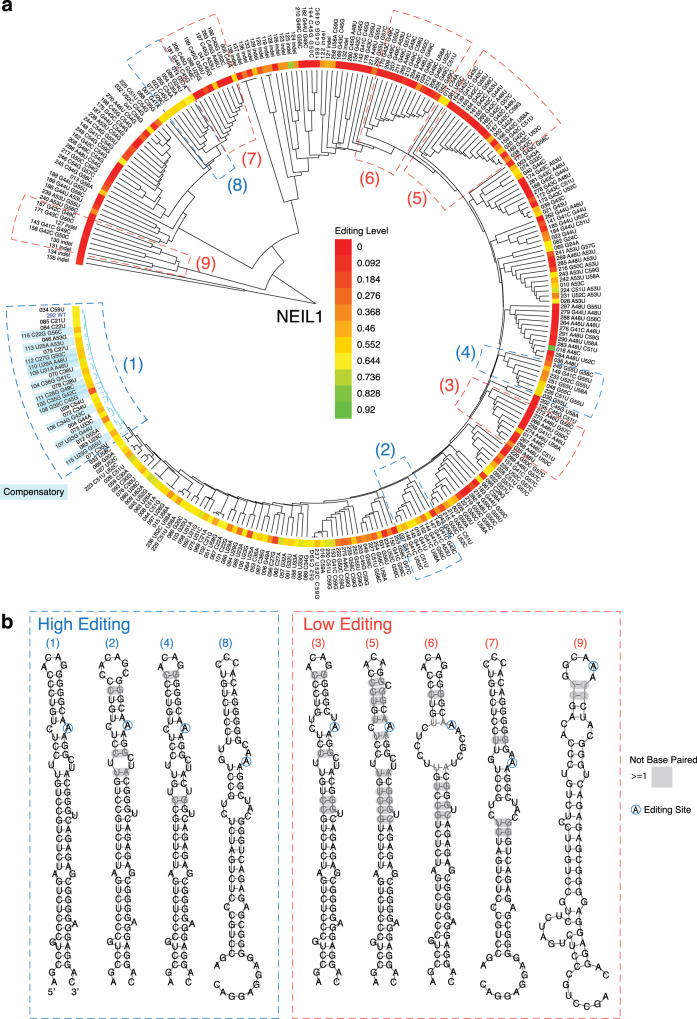
Adenosine-to-inosine (A-to-I) RNA editing catalyzed by ADAR enzymes occurs in double-stranded RNAs. Despite a compelling need towards predictive understanding of natural and engineered editing events, how the RNA sequence and structure determine the editing efficiency and specificity (i.e., cis-regulation) is poorly understood. We apply a CRISPR/Cas9-mediated saturation mutagenesis approach to generate libraries of mutations near three natural editing substrates at their endogenous genomic loci. We use machine learning to integrate diverse RNA sequence and structure features to model editing levels measured by deep sequencing. We confirm known features and identify new features important for RNA editing. Training and testing XGBoost algorithm within the same substrate yield models that explain 68 to 86 percent of substrate-specific variation in editing levels. However, the models do not generalize across substrates, suggesting complex and context-dependent regulation patterns. Our integrative approach can be applied to larger scale experiments towards deciphering the RNA editing code.
Conflict of interest statement
The authors declare the following competing interests: J.B.L. is a co-founder of AIRNA Bio and a consultant for Risen Pharma. Anna Shcherbina receives consulting fees from Myokardia, Inc, is a scientific adviser to Ravel Bio, Inc., and an employee of Insitro, Inc.
Figures



Similar articles
-
Current strategies for Site-Directed RNA Editing using ADARs.Methods. 2019 Mar 1;156:16-24. doi: 10.1016/j.ymeth.2018.11.016. Epub 2018 Nov 29. Methods. 2019. PMID: 30502398 Free PMC article. Review.
-
Use of CRISPR/Cas Genome Editing Technology for Targeted Mutagenesis in Rice.Methods Mol Biol. 2017;1498:33-40. doi: 10.1007/978-1-4939-6472-7_3. Methods Mol Biol. 2017. PMID: 27709567
-
Adenosine to inosine editing by ADAR2 requires formation of a ternary complex on the GluR-B R/G site.J Biol Chem. 2002 Oct 4;277(40):37624-9. doi: 10.1074/jbc.M204126200. Epub 2002 Aug 5. J Biol Chem. 2002. PMID: 12163487
-
High-throughput screening for functional adenosine to inosine RNA editing systems.ACS Chem Biol. 2006 Dec 15;1(12):761-5. doi: 10.1021/cb6003838. ACS Chem Biol. 2006. PMID: 17240974
-
A-to-I editing challenger or ally to the microRNA process.Biochimie. 2007 Oct;89(10):1171-6. doi: 10.1016/j.biochi.2007.06.002. Epub 2007 Jun 8. Biochimie. 2007. PMID: 17628290 Review.
Cited by
-
A systematic mapping study on machine learning techniques for the prediction of CRISPR/Cas9 sgRNA target cleavage.Comput Struct Biotechnol J. 2022 Oct 21;20:5813-5823. doi: 10.1016/j.csbj.2022.10.013. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36382194 Free PMC article. Review.
-
RNA editing: Expanding the potential of RNA therapeutics.Mol Ther. 2023 Jun 7;31(6):1533-1549. doi: 10.1016/j.ymthe.2023.01.005. Epub 2023 Jan 7. Mol Ther. 2023. PMID: 36620962 Free PMC article. Review.
-
Multiplexed assays of variant effect for clinical variant interpretation.Nat Rev Genet. 2025 Jul 21. doi: 10.1038/s41576-025-00870-x. Online ahead of print. Nat Rev Genet. 2025. PMID: 40691352 Review.
-
Precise in vivo RNA base editing with a wobble-enhanced circular CLUSTER guide RNA.Nat Biotechnol. 2025 Apr;43(4):545-557. doi: 10.1038/s41587-024-02313-0. Epub 2024 Jul 12. Nat Biotechnol. 2025. PMID: 38997581 Free PMC article.
-
DEMINING: A deep learning model embedded framework to distinguish RNA editing from DNA mutations in RNA sequencing data.Genome Biol. 2024 Oct 8;25(1):258. doi: 10.1186/s13059-024-03397-2. Genome Biol. 2024. PMID: 39380061 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
