

Computing inelastic neutron scattering spectra from molecular dynamics trajectories
- PMID: 33846390
- PMCID: PMC8041884
- DOI: 10.1038/s41598-021-86771-5
Computing inelastic neutron scattering spectra from molecular dynamics trajectories
Abstract
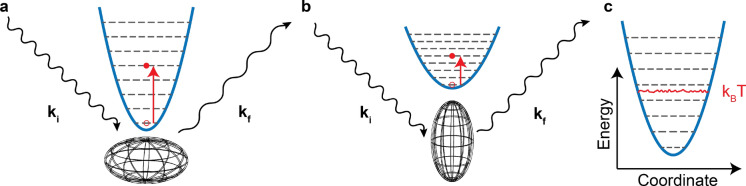
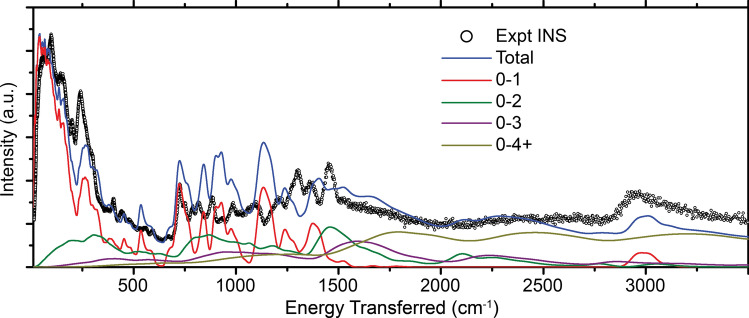
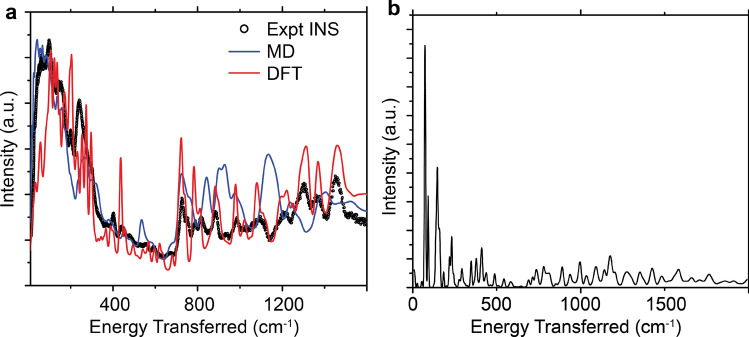
Inelastic neutron scattering (INS) provides a weighted density of phonon modes. Currently, INS spectra can only be interpreted for perfectly crystalline materials because of high computational cost for electronic simulations. INS has the potential to provide detailed morphological information if sufficiently large volumes and appropriate structural variety are simulated. Here, we propose a method that allows direct comparison between INS data with molecular dynamics simulations, a simulation method that is frequently used to simulate semicrystalline/amorphous materials. We illustrate the technique by analyzing spectra of a well-studied conjugated polymer, poly(3-hexylthiophene-2,5-diyl) (P3HT) and conclude that our technique provides improved volume and structural variety, but that the classical force field requires improvement before the morphology can be accurately interpreted.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Crabtree, G., Glotzer, S., McCurdy, B. & Roberto, J. Computational materials science and chemistry: Accelerating discovery and innovation through simulation-based engineering and science. Report of the US Department of Energy Workshop on Computational Materials Science and Chemistry for Innovation (2010).
-
- Bousige, C. et al. Realistic molecular model of kerogen’s nanostructure. Nat. Mater.15, 576–582 (2016). - PubMed
-
- Poelking C, Andrienko D. Effect of polymorphism, regioregularity and paracrystallinity on charge transport in poly(3-hexylthiophene) [P3HT] nanofibers. Macromolecules. 2013;46:8941–8956. doi: 10.1021/ma4015966. - DOI
-
- Harrelson TF, et al. Identifying atomic scale structure in undoped/doped semicrystalline P3HT using inelastic neutron scattering. Macromolecules. 2017;50:2424–2435. doi: 10.1021/acs.macromol.6b02410. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
