

Integration of genomic sequencing into the response to the Ebola virus outbreak in Nord Kivu, Democratic Republic of the Congo
- PMID: 33846610
- PMCID: PMC8549801
- DOI: 10.1038/s41591-021-01302-z
Integration of genomic sequencing into the response to the Ebola virus outbreak in Nord Kivu, Democratic Republic of the Congo
Abstract
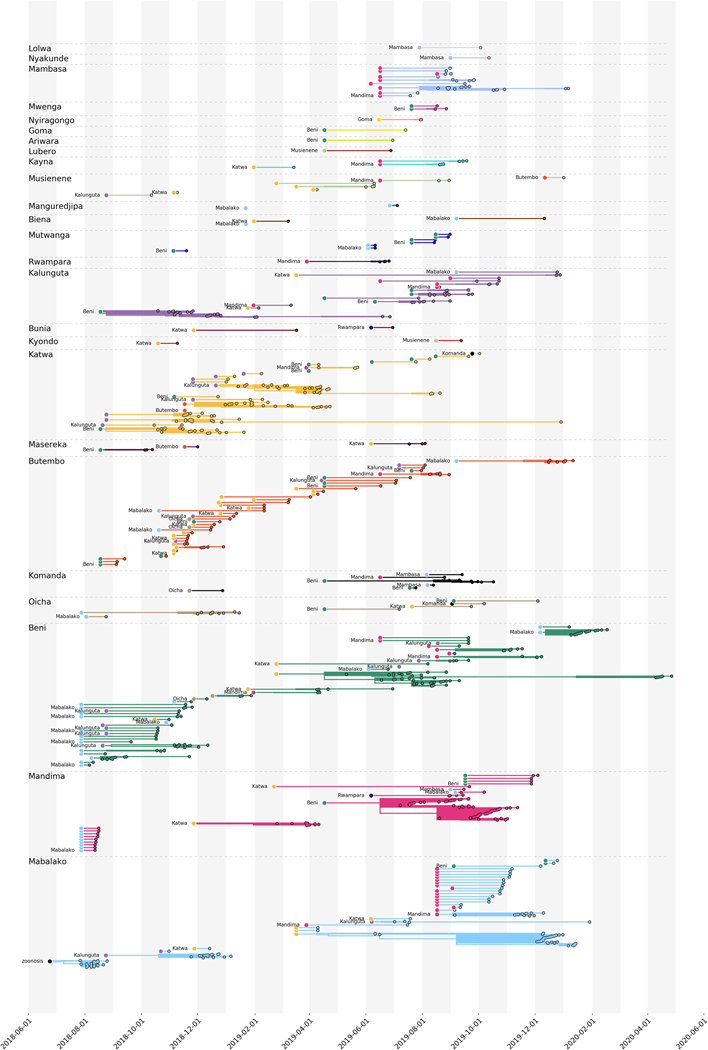
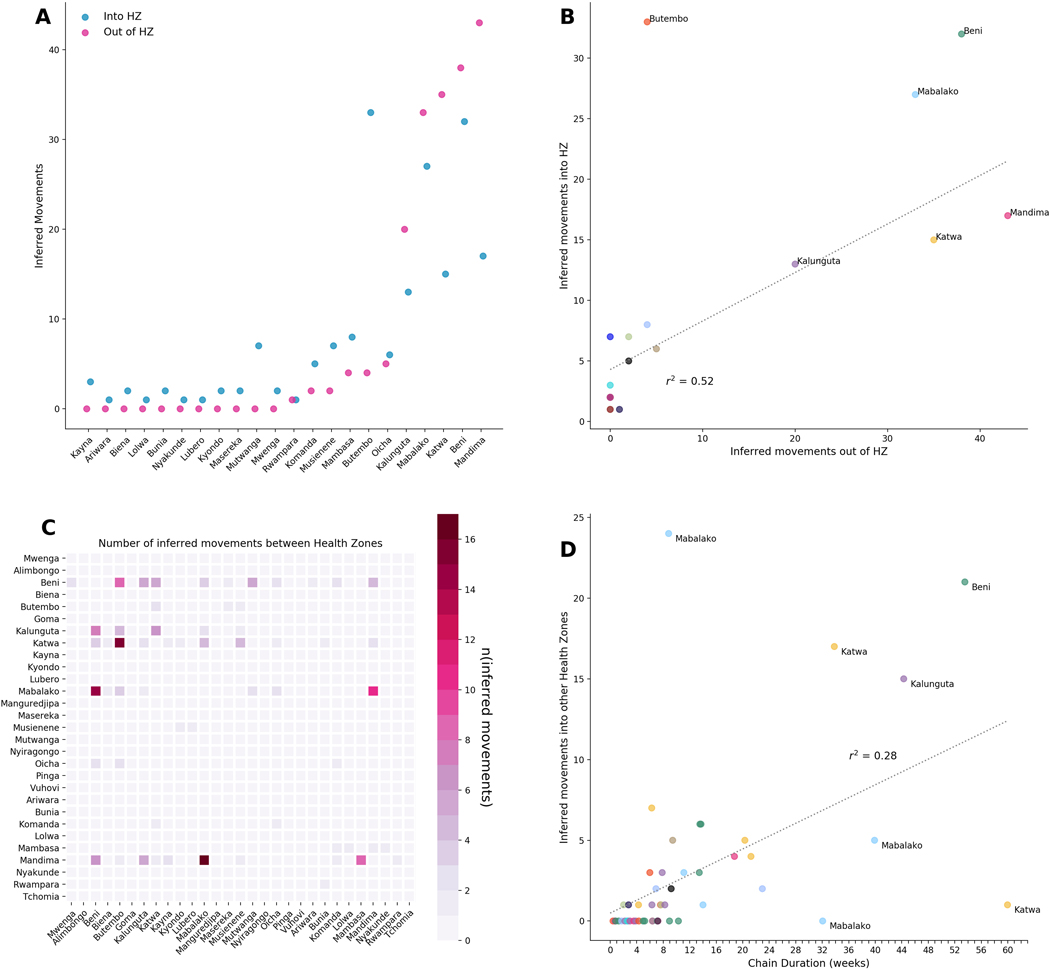
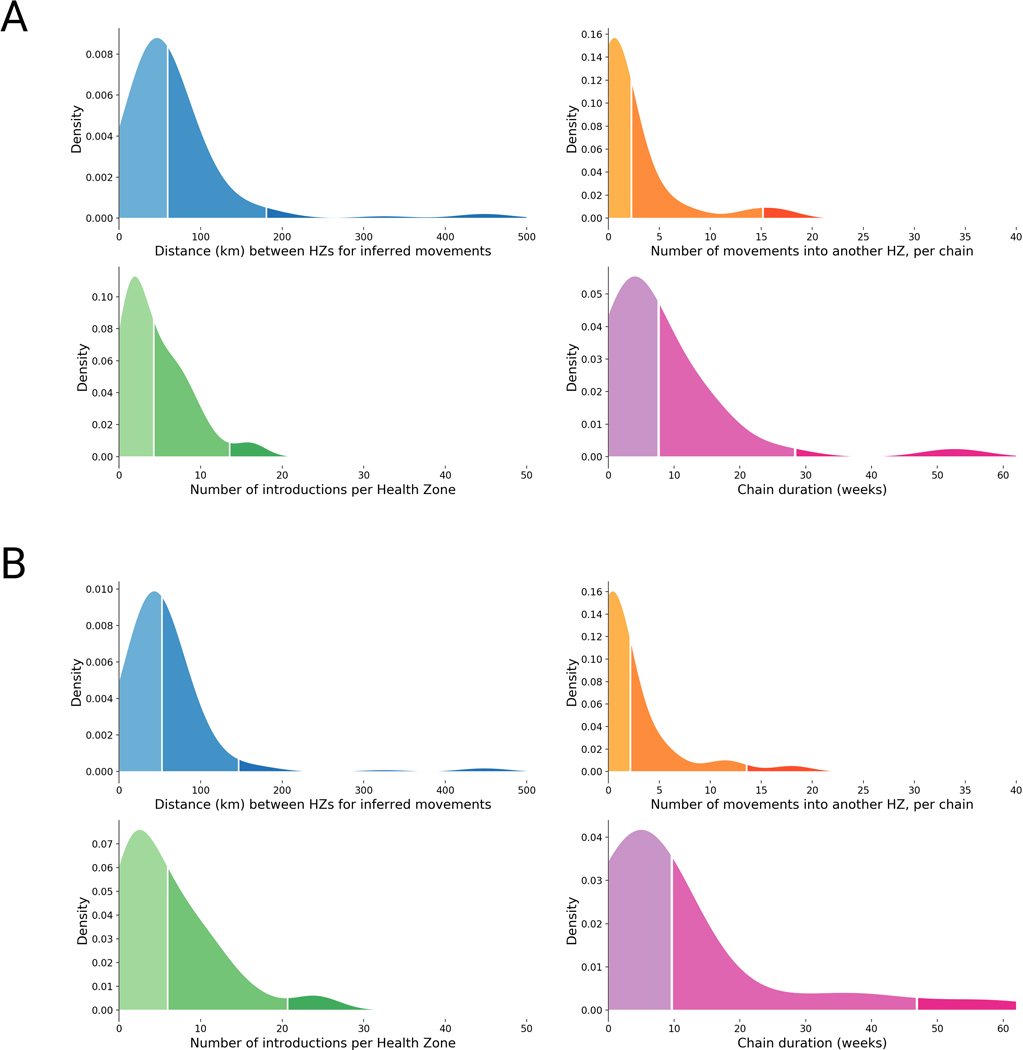
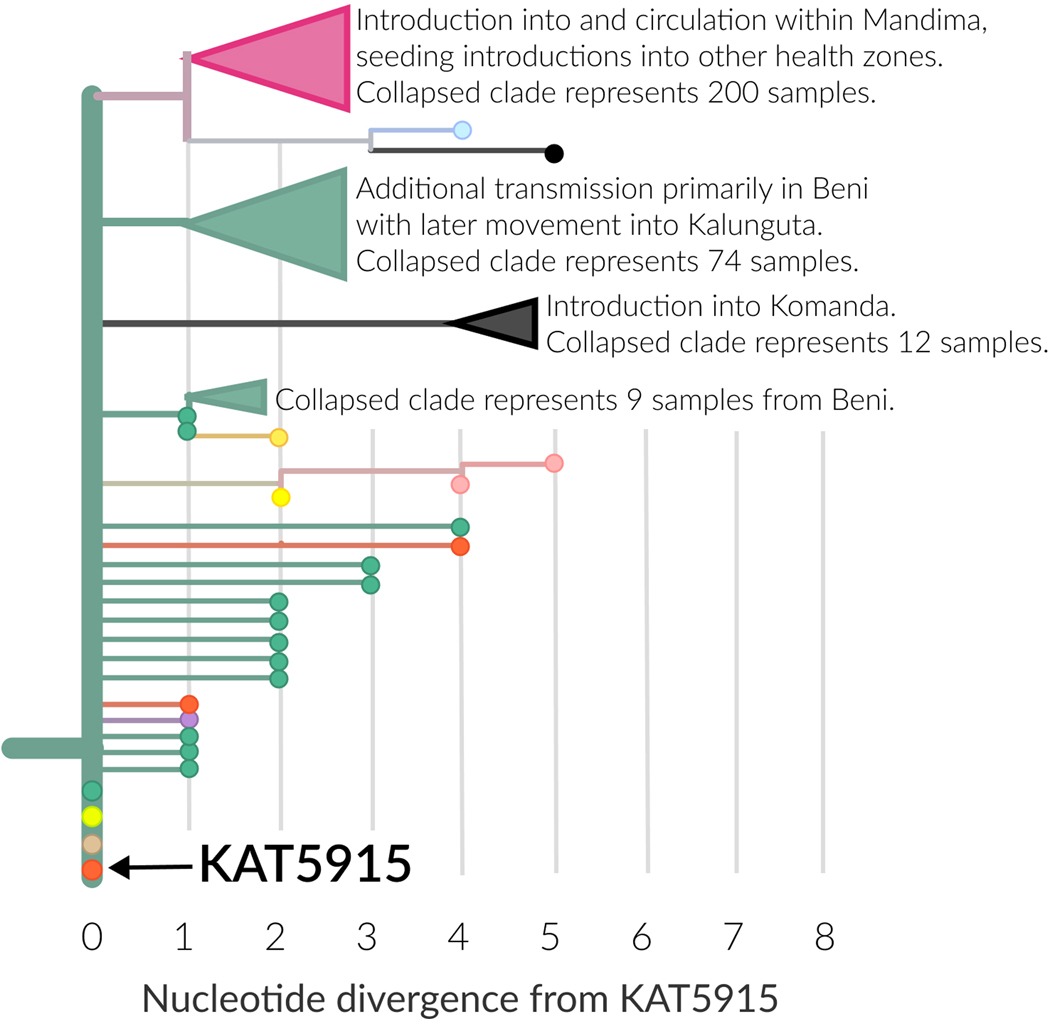
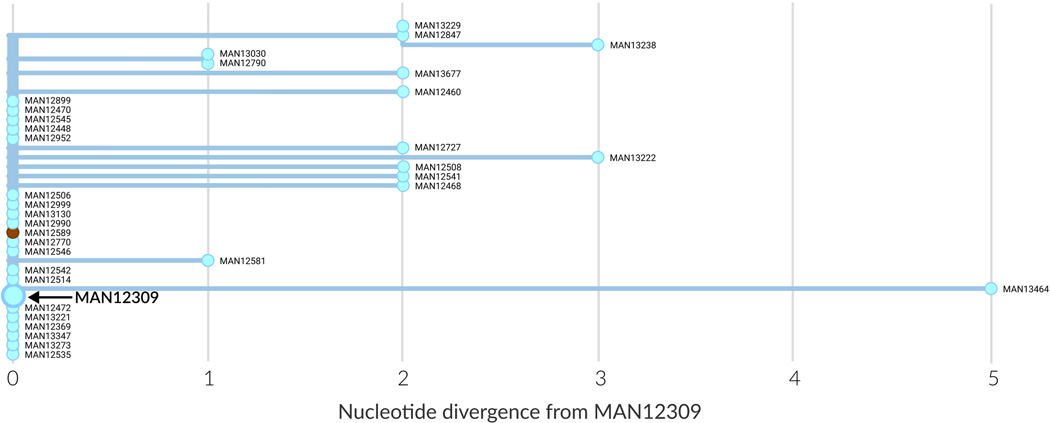
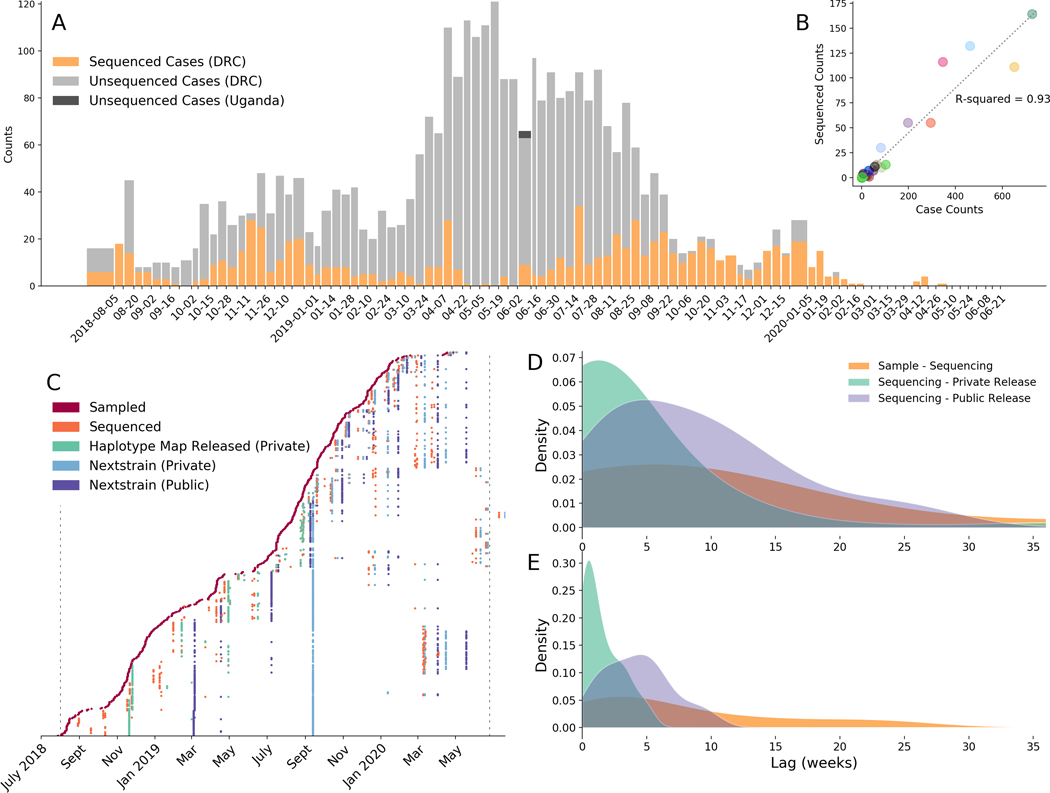
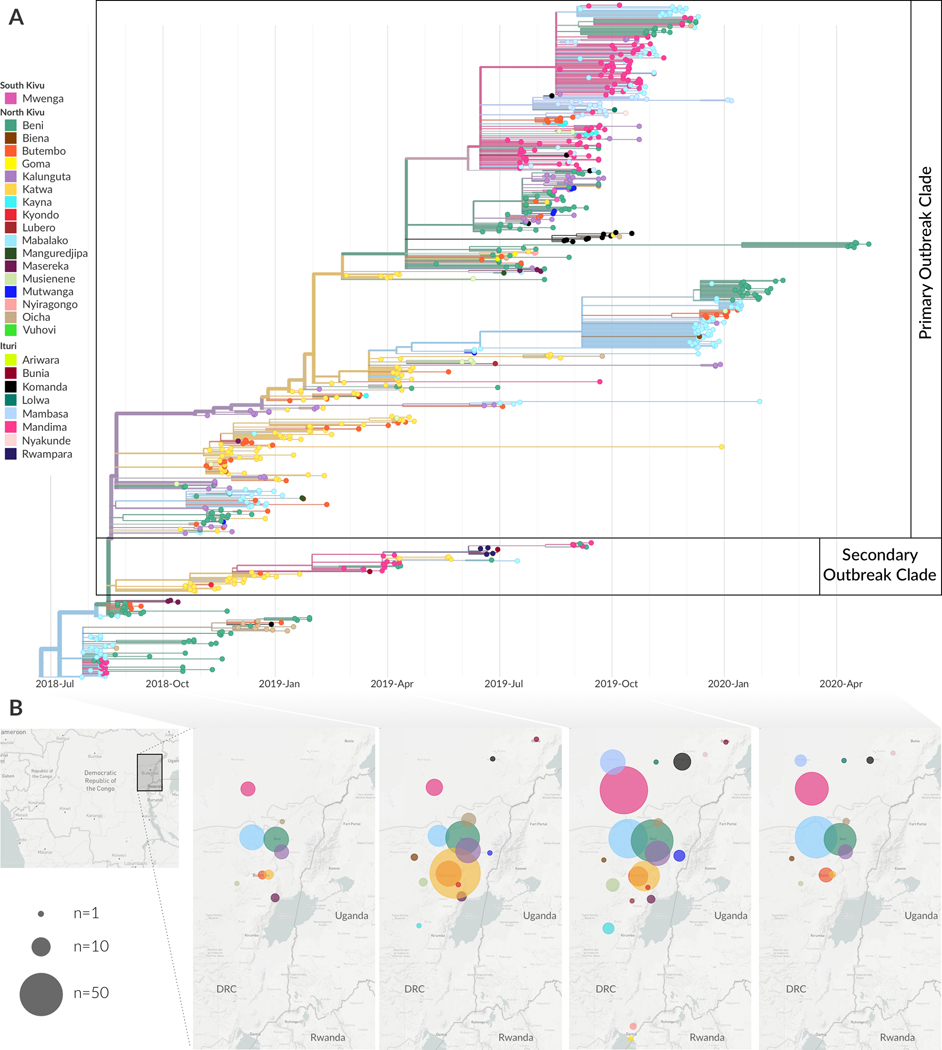
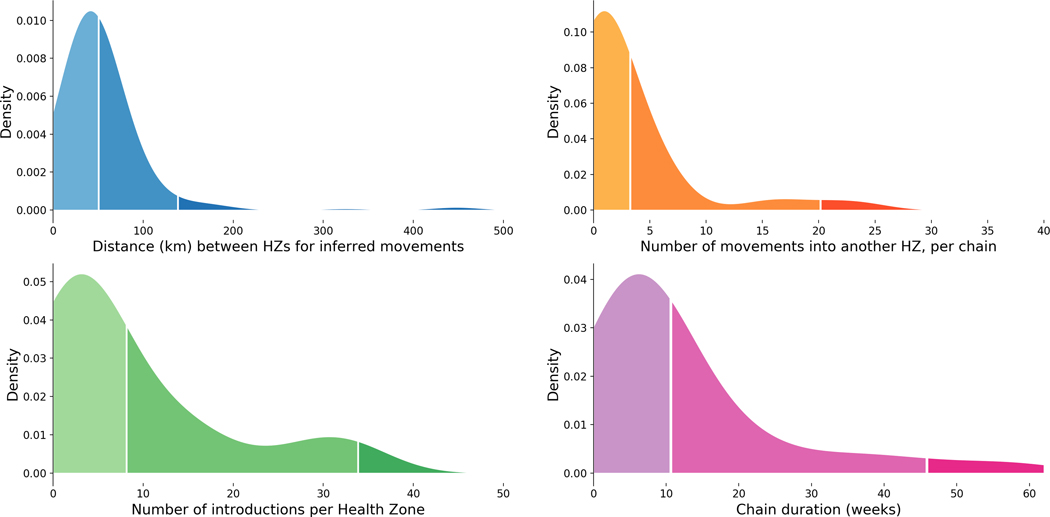
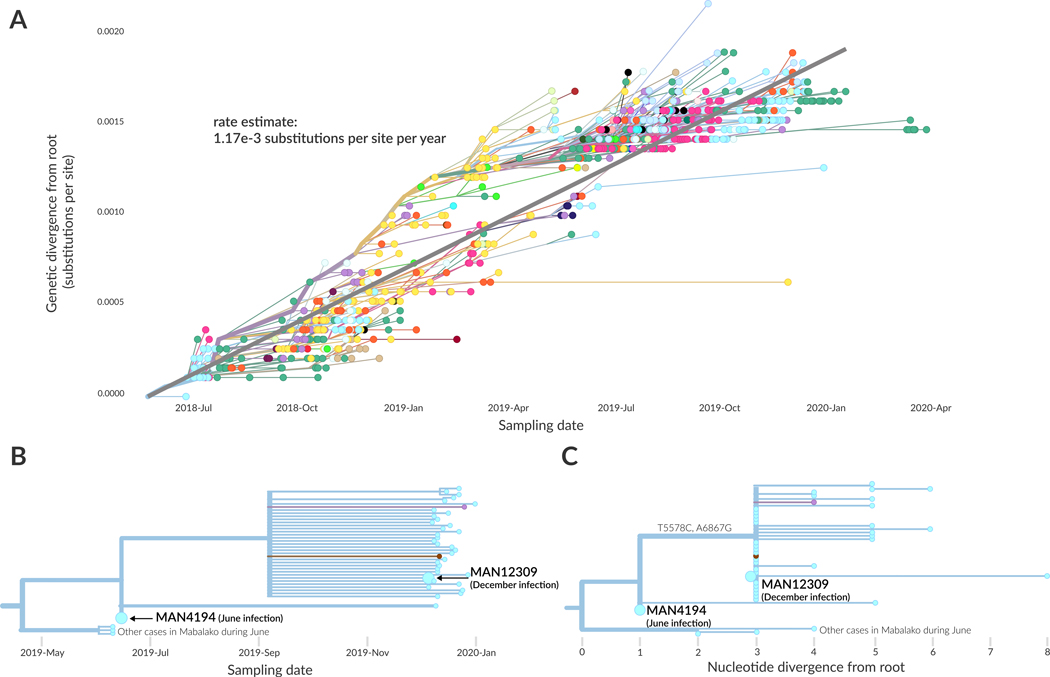
On 1 August 2018, the Democratic Republic of the Congo (DRC) declared its tenth Ebola virus disease (EVD) outbreak. To aid the epidemiologic response, the Institut National de Recherche Biomédicale (INRB) implemented an end-to-end genomic surveillance system, including sequencing, bioinformatic analysis and dissemination of genomic epidemiologic results to frontline public health workers. We report 744 new genomes sampled between 27 July 2018 and 27 April 2020 generated by this surveillance effort. Together with previously available sequence data (n = 48 genomes), these data represent almost 24% of all laboratory-confirmed Ebola virus (EBOV) infections in DRC in the period analyzed. We inferred spatiotemporal transmission dynamics from the genomic data as new sequences were generated, and disseminated the results to support epidemiologic response efforts. Here we provide an overview of how this genomic surveillance system functioned, present a full phylodynamic analysis of 792 Ebola genomes from the Nord Kivu outbreak and discuss how the genomic surveillance data informed response efforts and public health decision making.
Conflict of interest statement
Competing Interests Statement
The authors declare no competing interests.
Figures
References
Methods-only References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
