

Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology
- PMID: 33846644
- PMCID: PMC8434938
- DOI: 10.1038/s41587-021-00857-z
Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology
Abstract
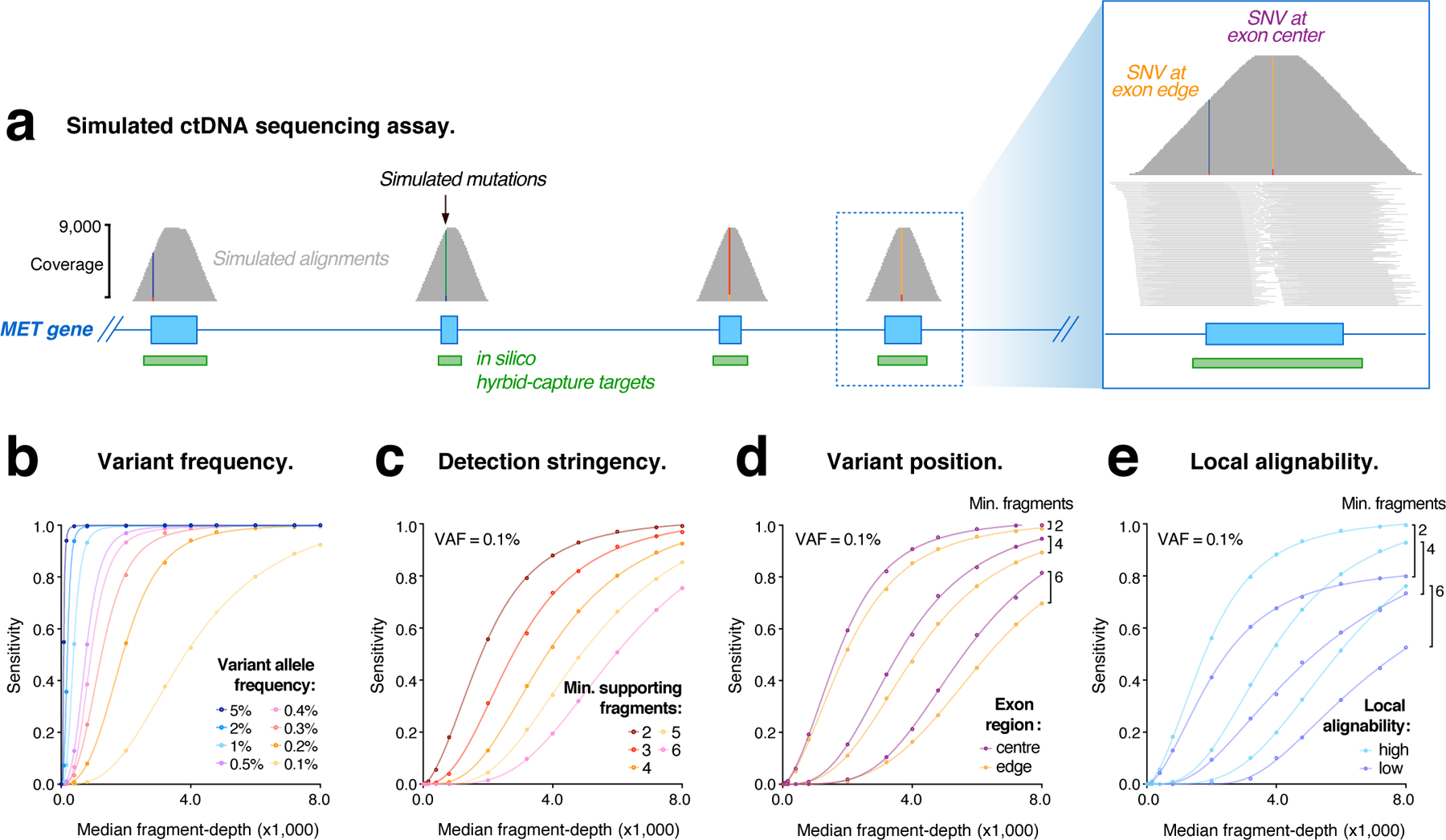
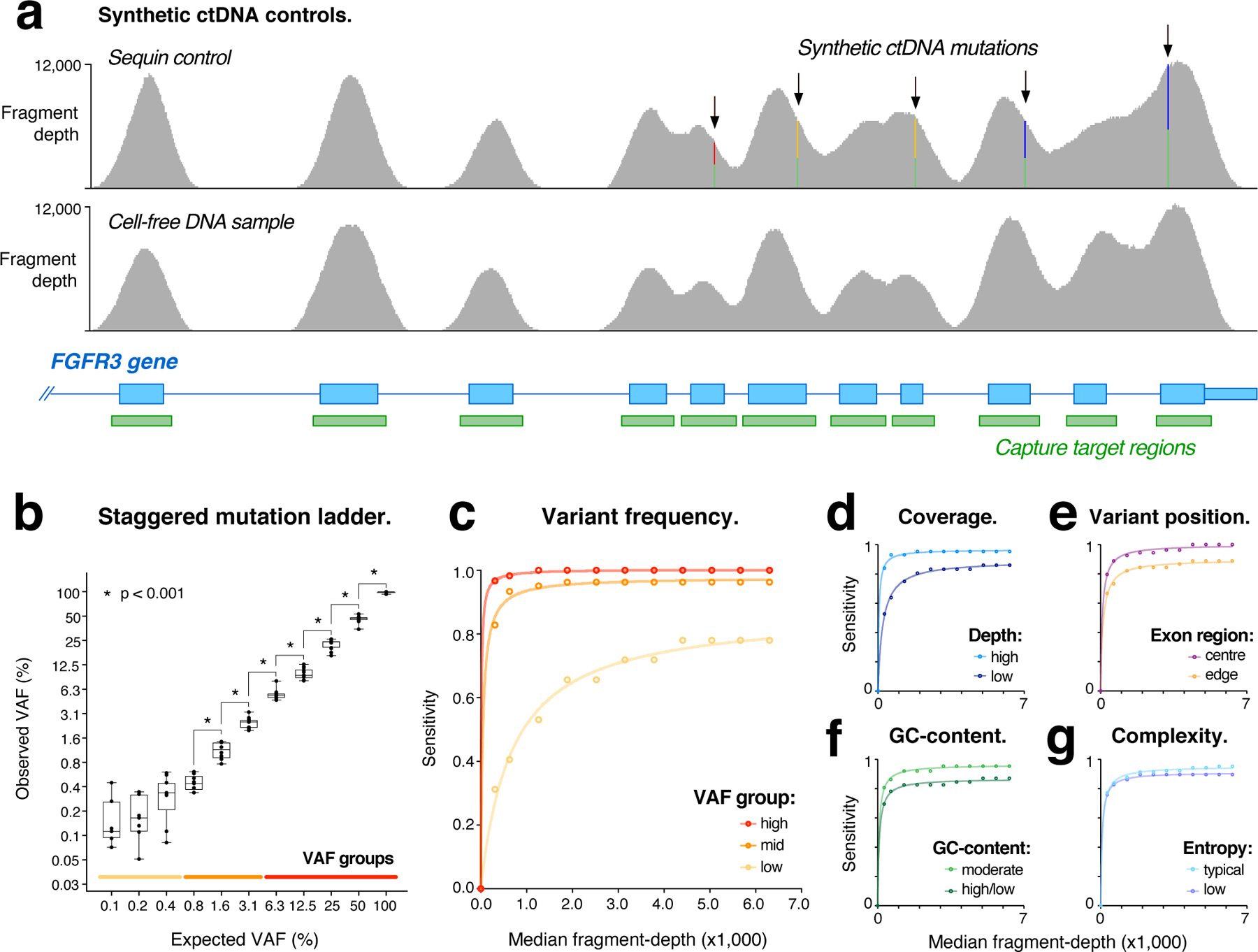
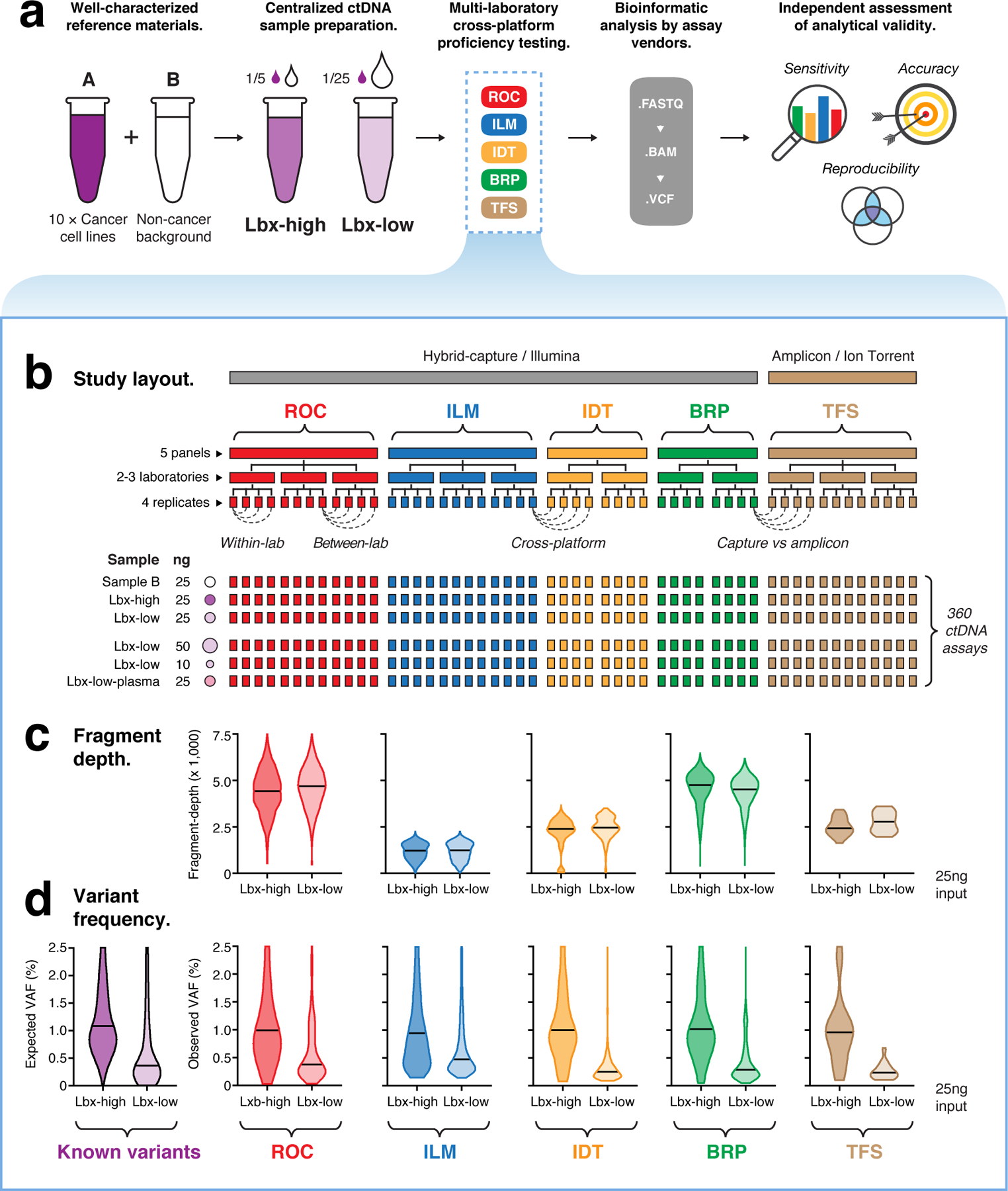
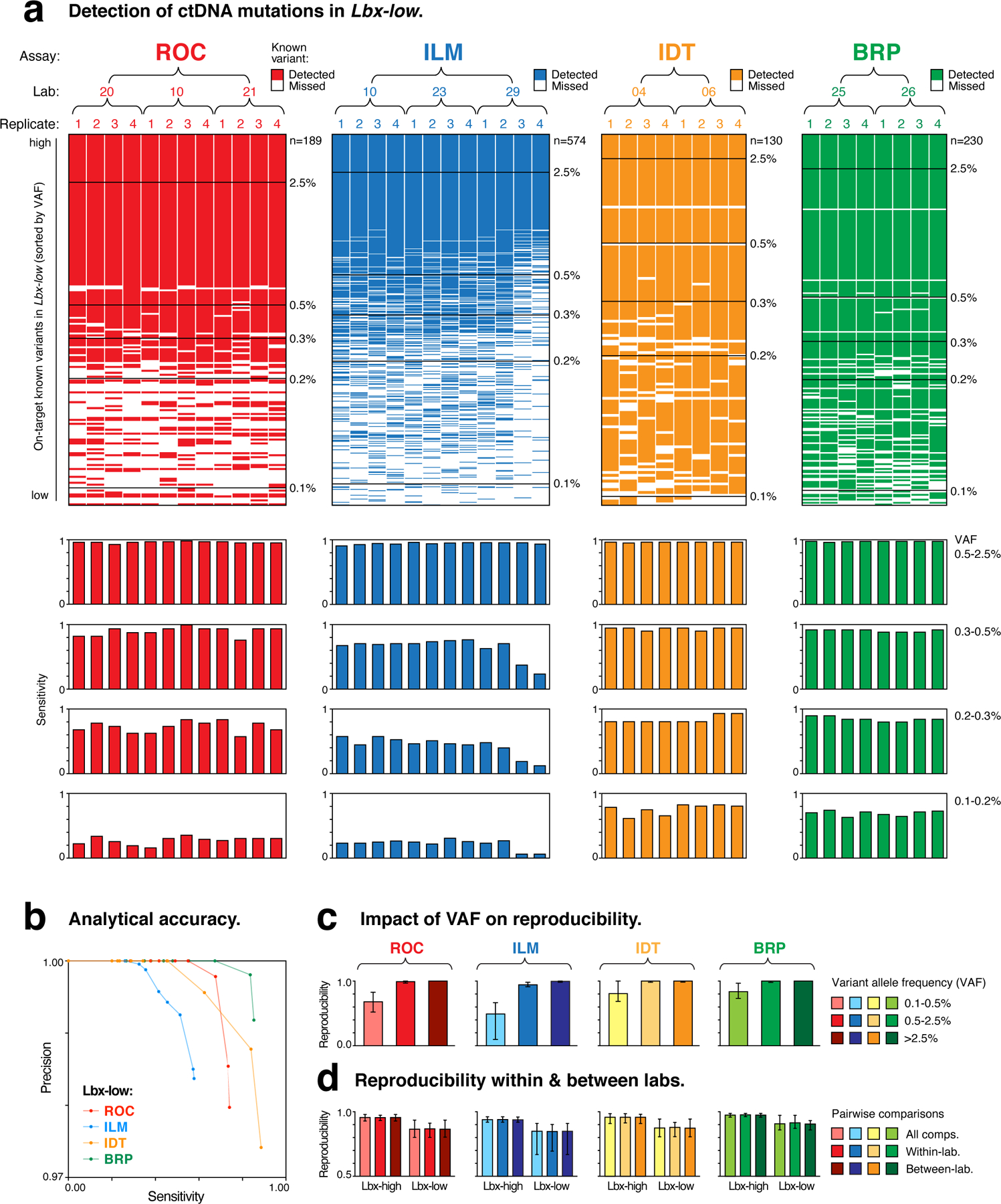
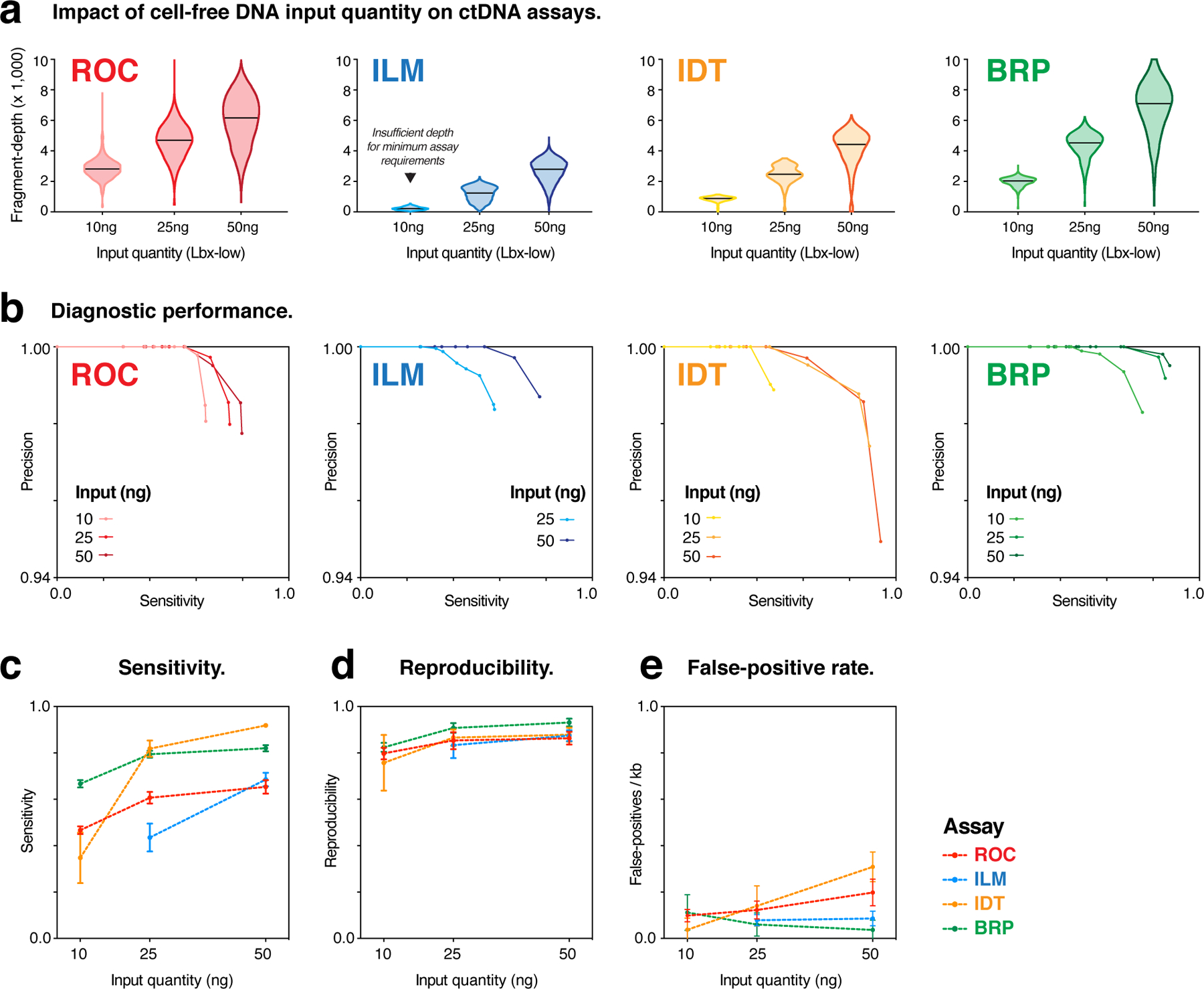
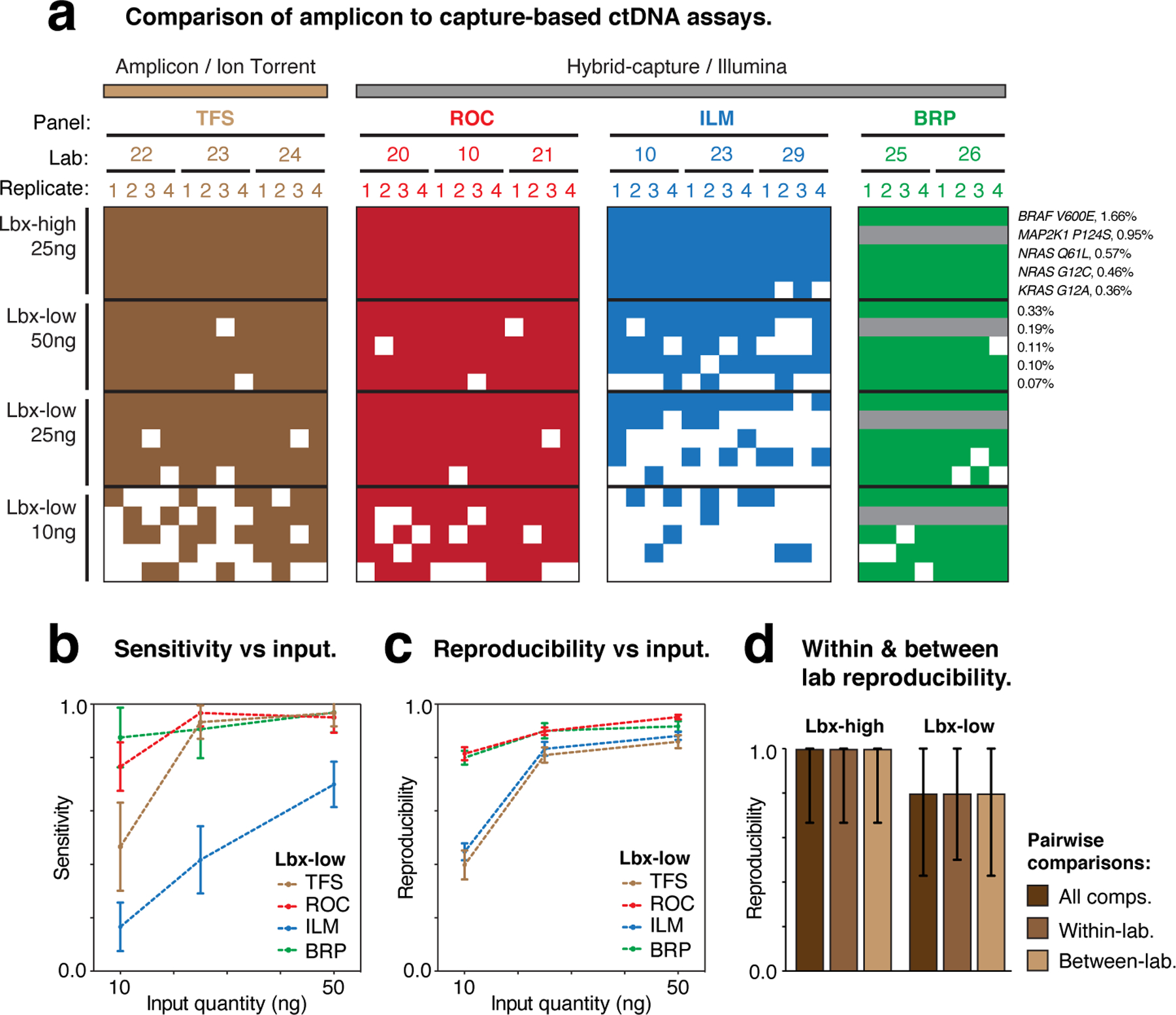
Circulating tumor DNA (ctDNA) sequencing is being rapidly adopted in precision oncology, but the accuracy, sensitivity and reproducibility of ctDNA assays is poorly understood. Here we report the findings of a multi-site, cross-platform evaluation of the analytical performance of five industry-leading ctDNA assays. We evaluated each stage of the ctDNA sequencing workflow with simulations, synthetic DNA spike-in experiments and proficiency testing on standardized, cell-line-derived reference samples. Above 0.5% variant allele frequency, ctDNA mutations were detected with high sensitivity, precision and reproducibility by all five assays, whereas, below this limit, detection became unreliable and varied widely between assays, especially when input material was limited. Missed mutations (false negatives) were more common than erroneous candidates (false positives), indicating that the reliable sampling of rare ctDNA fragments is the key challenge for ctDNA assays. This comprehensive evaluation of the analytical performance of ctDNA assays serves to inform best practice guidelines and provides a resource for precision oncology.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
DISCLAIMERS & COMPETING INTERESTS
This research includes contributions from, and was reviewed by, the FDA and the NIH. This work has been approved for publication by these agencies, but it does not necessarily reflect official agency policy. Certain commercial materials and equipment are identified in order to adequately specify experimental procedures. In no case does such identification imply recommendation or endorsement by the FDA or the NIH, nor does it imply that the items identified are necessarily the best available for purpose. The Garvan Institute of Medical Research has filed patent applications on synthetic controls for genomics. The authors declare no other competing financial interests.
Figures
References
-
- Leon SA, Shapiro B, Sklaroff DM & Yaros MJ Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 37, 646–650 (1977). - PubMed
-
- Stroun M, Anker P, Lyautey J, Lederrey C & Maurice PA Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol 23, 707–712 (1987). - PubMed
-
- Abbosh C, Birkbak NJ & Swanton C Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nat Rev Clin Oncol 15, 577–586 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
