

Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues
- PMID: 33846645
- PMCID: PMC7611252
- DOI: 10.1038/s41587-021-00869-9
Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues
Abstract
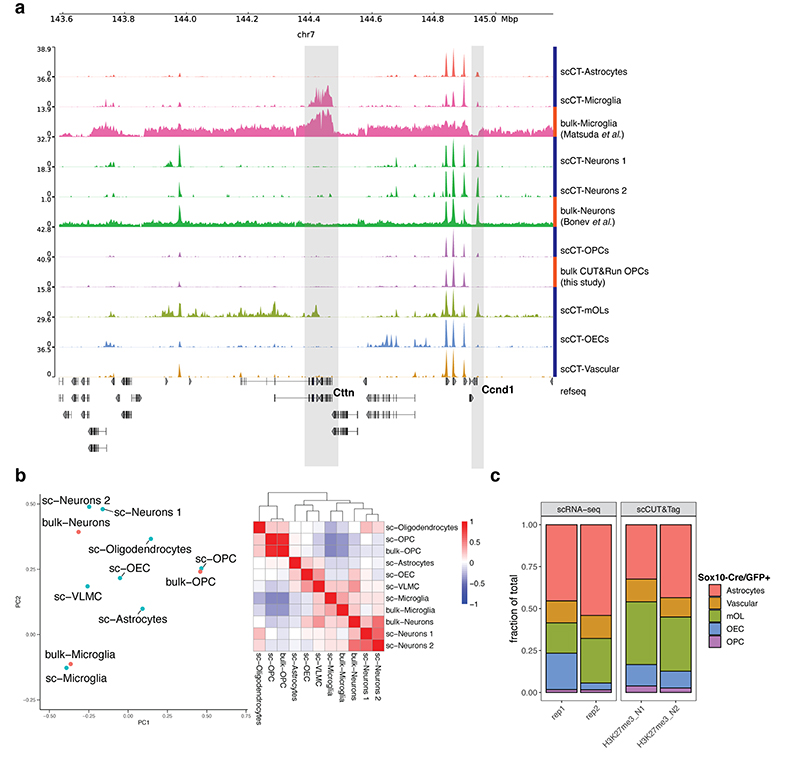
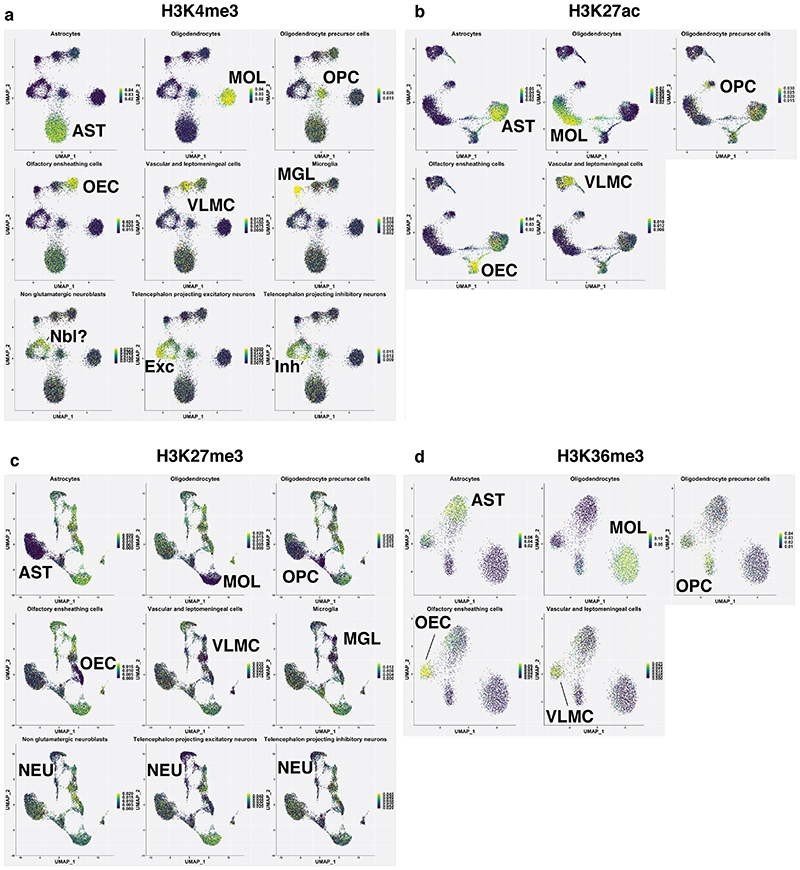
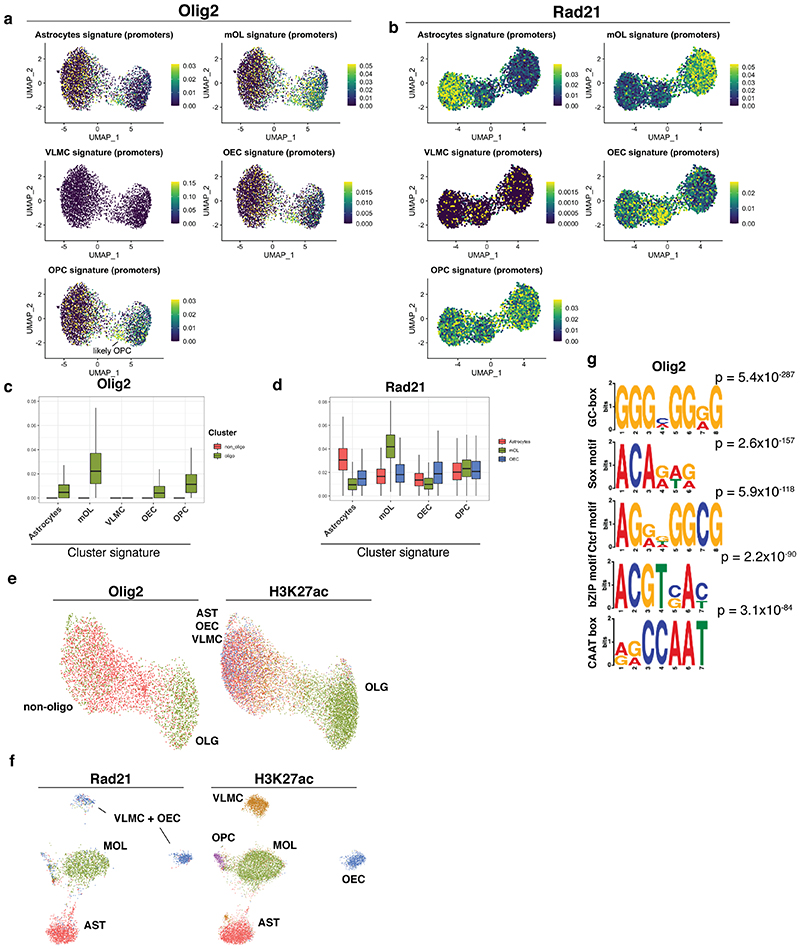
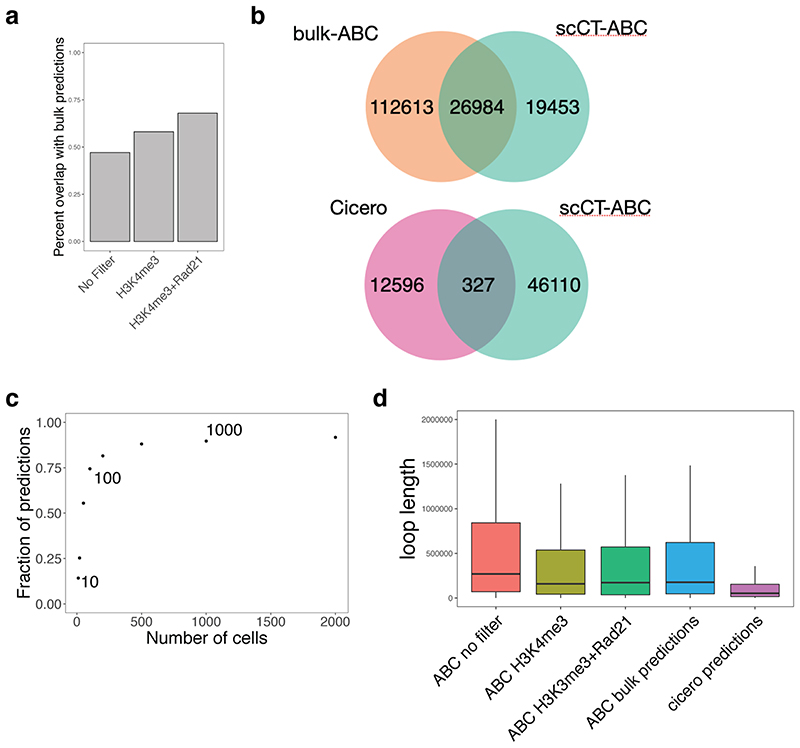
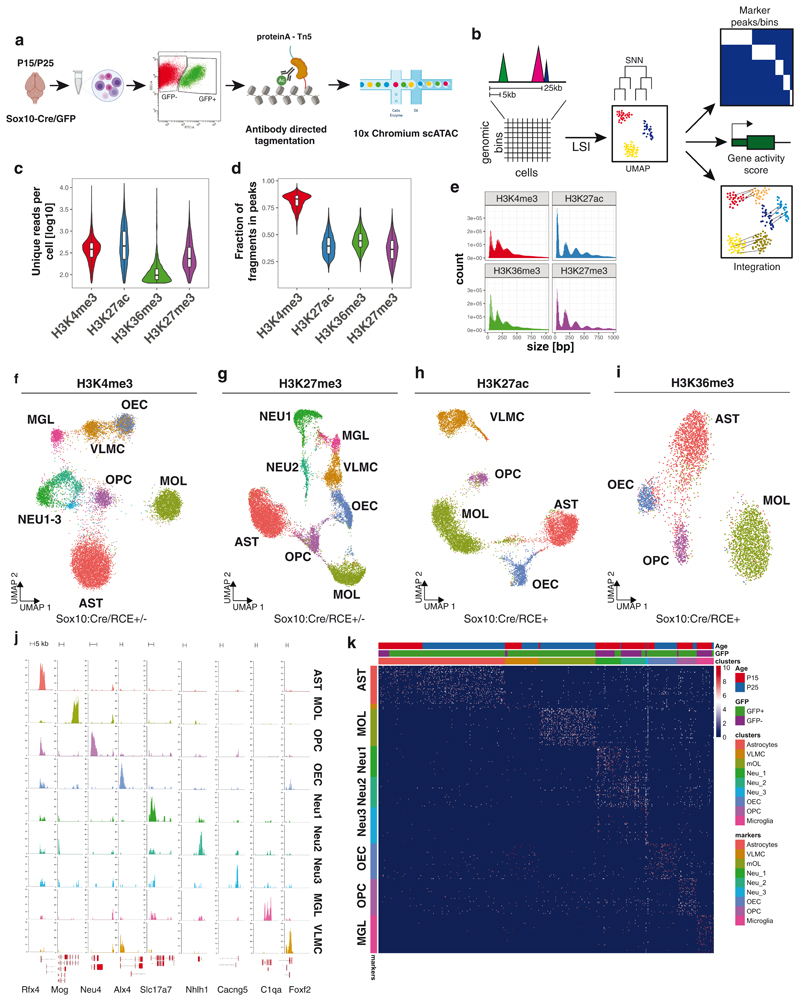
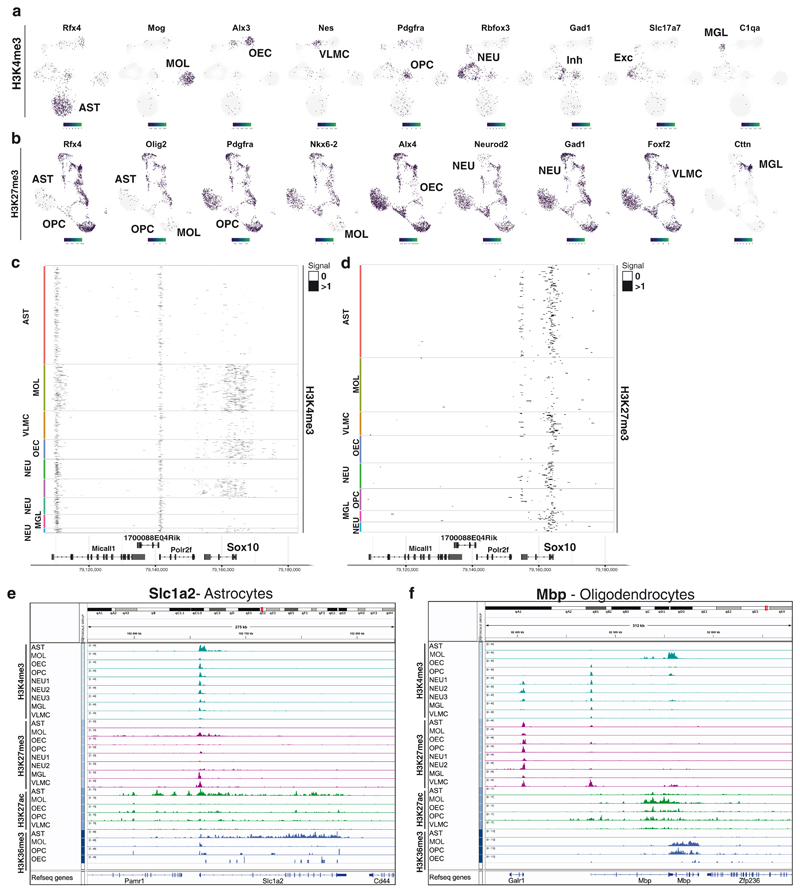
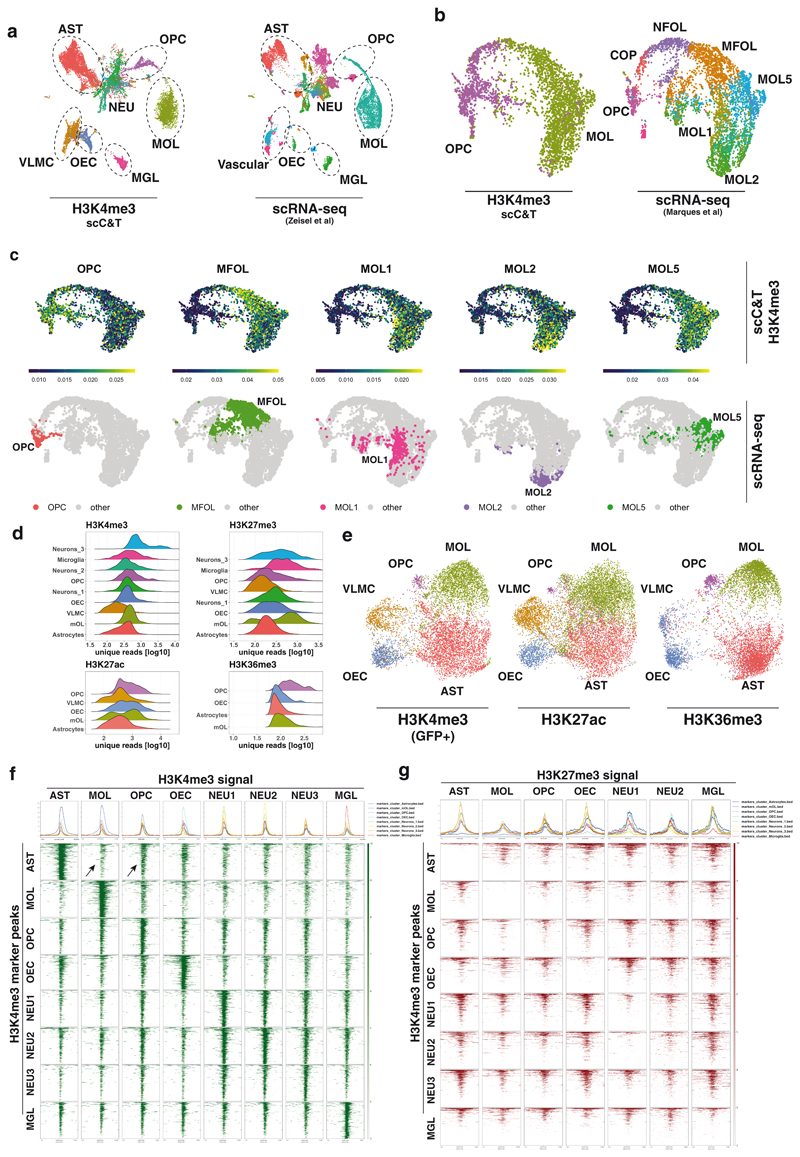
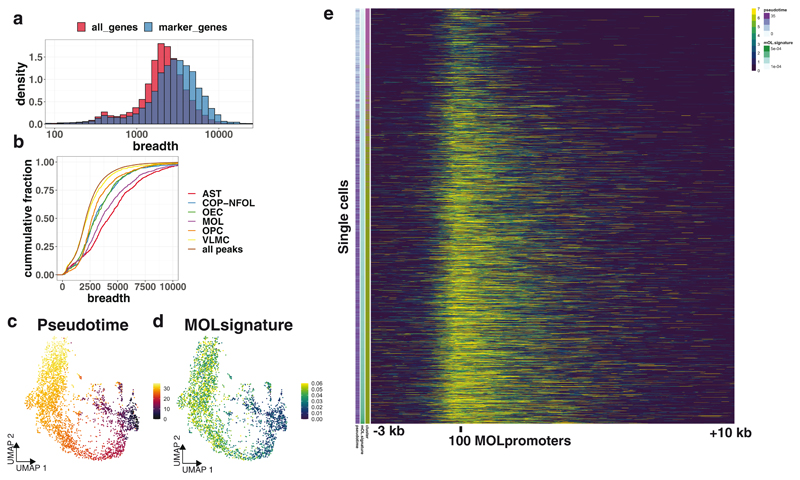
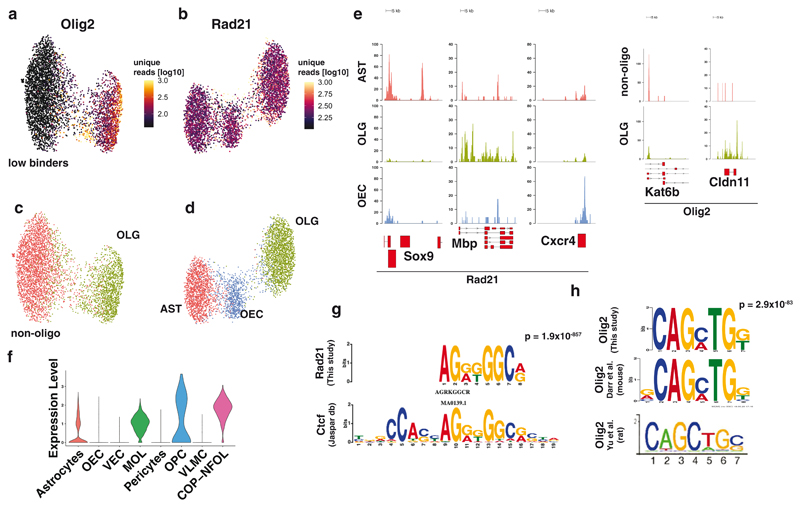
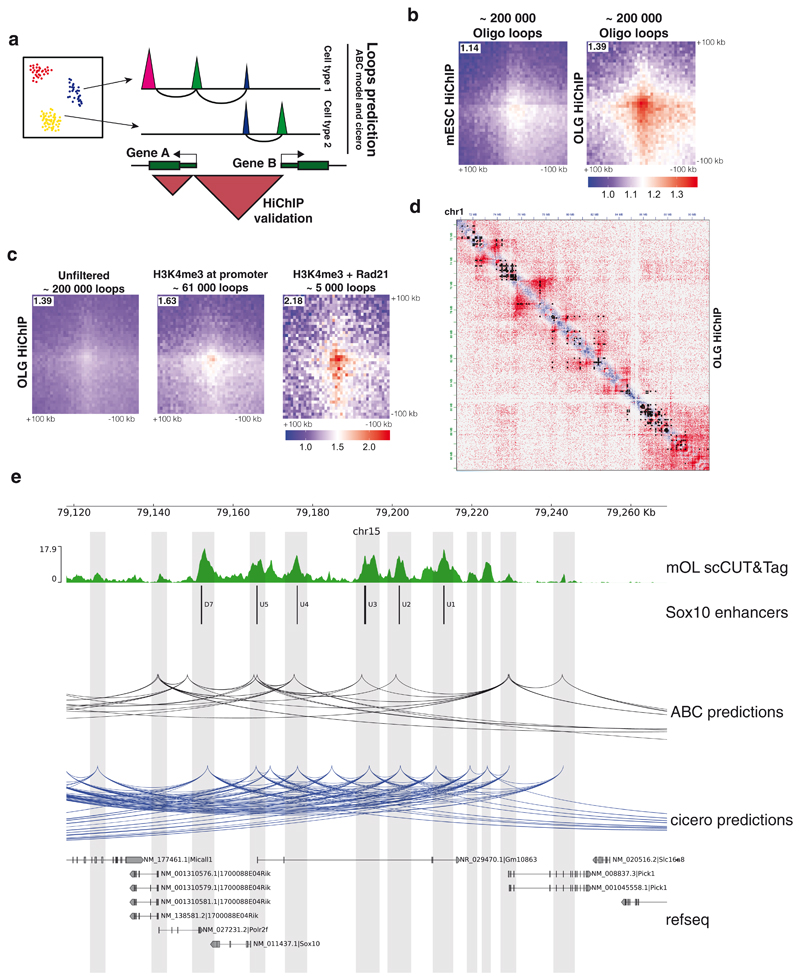
In contrast to single-cell approaches for measuring gene expression and DNA accessibility, single-cell methods for analyzing histone modifications are limited by low sensitivity and throughput. Here, we combine the CUT&Tag technology, developed to measure bulk histone modifications, with droplet-based single-cell library preparation to produce high-quality single-cell data on chromatin modifications. We apply single-cell CUT&Tag (scCUT&Tag) to tens of thousands of cells of the mouse central nervous system and probe histone modifications characteristic of active promoters, enhancers and gene bodies (H3K4me3, H3K27ac and H3K36me3) and inactive regions (H3K27me3). These scCUT&Tag profiles were sufficient to determine cell identity and deconvolute regulatory principles such as promoter bivalency, spreading of H3K4me3 and promoter-enhancer connectivity. We also used scCUT&Tag to investigate the single-cell chromatin occupancy of transcription factor OLIG2 and the cohesin complex component RAD21. Our results indicate that analysis of histone modifications and transcription factor occupancy at single-cell resolution provides unique insights into epigenomic landscapes in the central nervous system.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
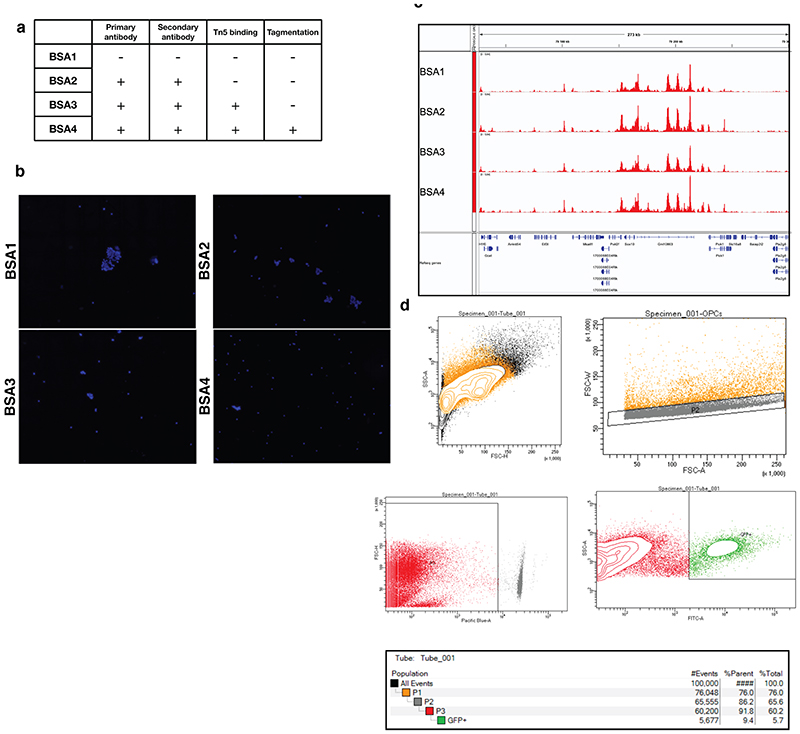
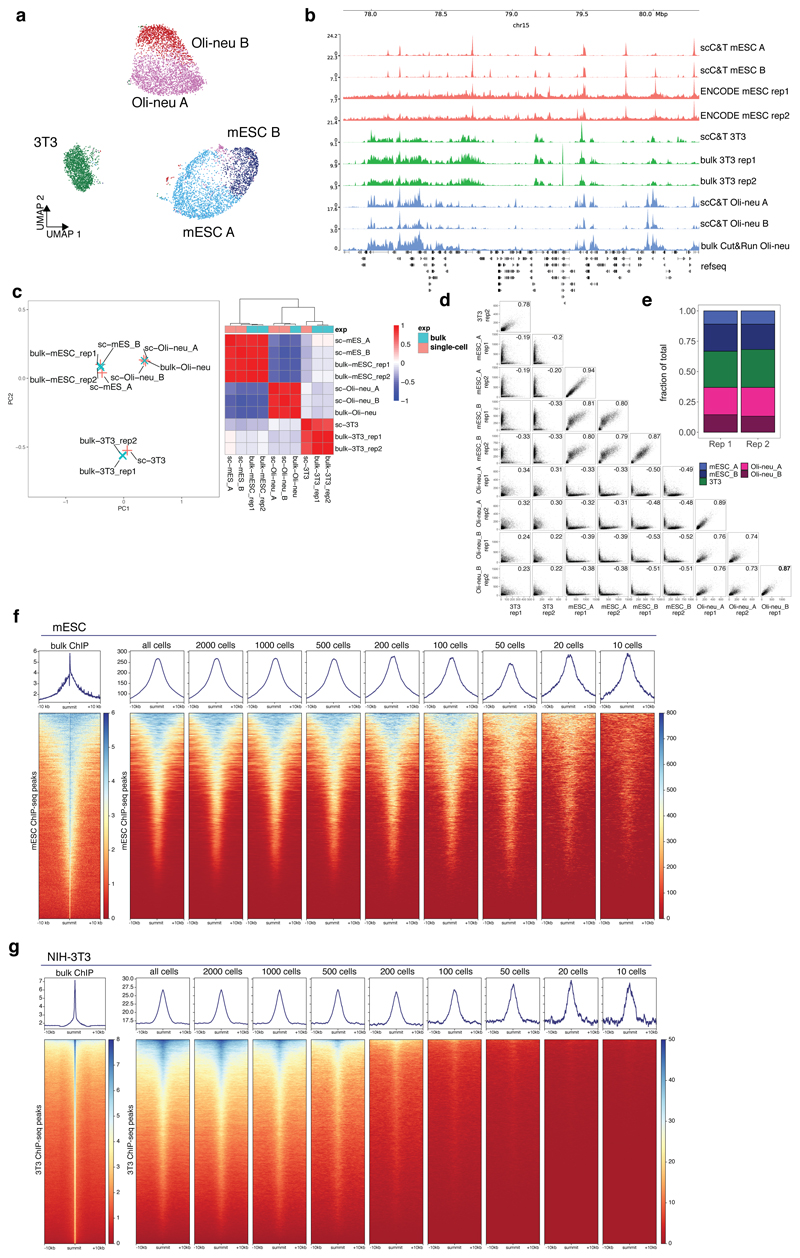
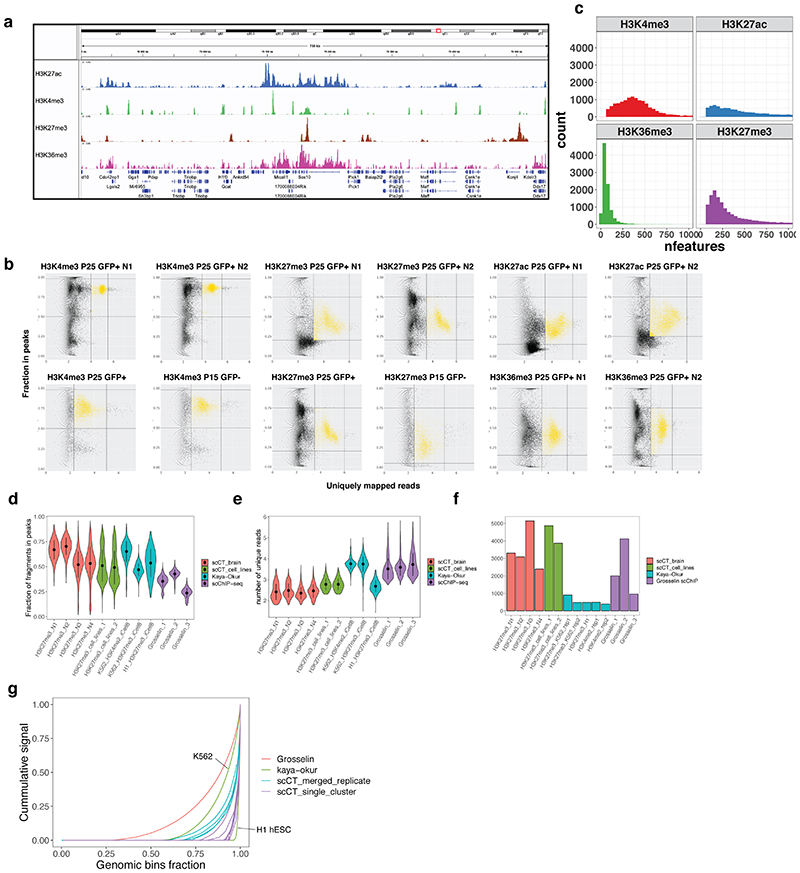

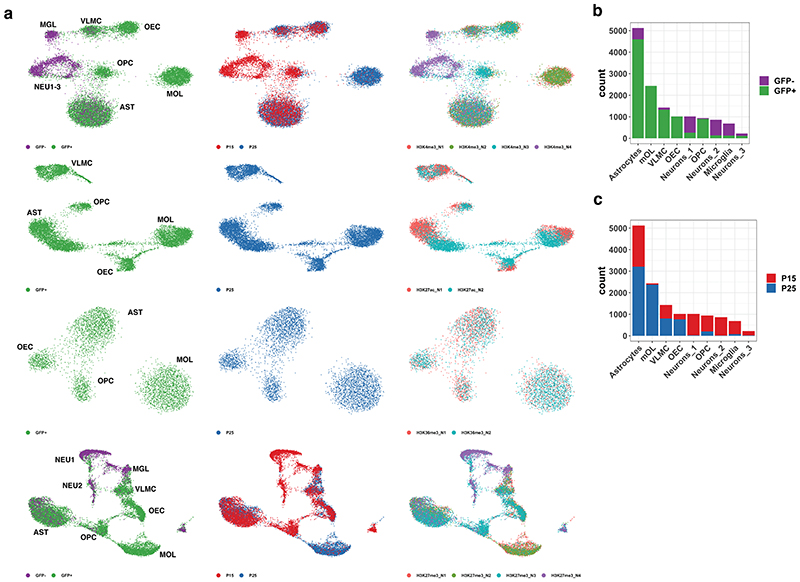

References
-
- Lorthongpanich C, et al. Single-Cell DNA-Methylation Analysis Reveals Epigenetic Chimerism in Preimplantation Embryos. Science. 2013;341:1110–1112. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
