

A novel KCNQ4 gene variant (c.857A>G; p.Tyr286Cys) in an extended family with non‑syndromic deafness 2A
- PMID: 33846771
- PMCID: PMC8025472
- DOI: 10.3892/mmr.2021.12059
A novel KCNQ4 gene variant (c.857A>G; p.Tyr286Cys) in an extended family with non‑syndromic deafness 2A
Abstract
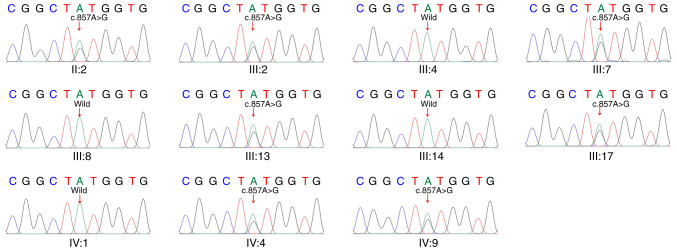
Deafness is one of the most common sensory disorders found in humans; notably, >60% of all cases of deafness have been attributed to genetic factors. Variants in potassium voltage‑gated channel subfamily Q member 4 (KCNQ4) are etiologically linked to a type of progressive hearing loss, deafness non‑syndromic autosomal dominant 2A (DFNA2A). In the present study, whole‑exome sequencing (WES) was performed on three members of a five‑generation Chinese family with 46 members with hearing loss. Pure tone audiometry and Sanger sequencing were performed for 11 family members to determine whether the novel variant in the KCNQ4 gene was segregated with the affected family members. In addition, evolutionary conservation analysis and computational tertiary structure protein prediction of the wild‑type KCNQ4 protein and its variant were performed. The family exhibited autosomal dominant, progressive, post‑lingual, non‑syndromic sensorineural hearing loss. A novel co‑segregating heterozygous missense variant (c.857A>G; p.Tyr286Cys) in the glycine‑tyrosine‑glycine signature sequence in the pore region of the KCNQ4 channel was identified. This variant was predicted to result in a tyrosine‑to‑cysteine substitution at position 286 in the KCNQ4 protein. The tyrosine at position 286 is well conserved across different species. The substitution of tyrosine with cysteine would affect the structure of the pore region, resulting in the loss of channel function. The KCNQ4 gene is one of the most common mutated genes observed in patients with autosomal dominant, non‑syndromic hearing loss. Taken together, for the family analyzed in the present study, performing WES in conjunction with Sanger sequencing has led to the detection of a novel, potentially causative variant (c.857 A>G; p.Tyr286Cys) in exon 6 of the KCNQ4 gene. The present study has added to the number of pathogenic variants observed in the KCNQ4 gene, and the findings may prove to be useful for both the diagnosis of DFNA2A and in the design of early interventional therapies.
Keywords: deafness; non‑syndromic autosomal dominant 2A; otassium voltage‑gated channel subfamily Q member 4; whole‑exome sequencing; glycine‑tyrosine‑glycine signature sequence; novel variant.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Zhang S, Zhang Y, Dong Y, Guo L, Zhang Z, Shao B, Qi J, Zhou H, Zhu W, Yan X, et al. Knockdown of Foxg1 in supporting cells increases the trans-differentiation of supporting cells into hair cells in the neonatal mouse cochlea. Cell Mol Life Sci. 2020;77:1401–1419. doi: 10.1007/s00018-019-03291-2. - DOI - PMC - PubMed
-
- Ralli M, Balla MP, Greco A, Altissimi G, Ricci P, Turchetta R, de Virgilio A, de Vincentiis M, Ricci S, Cianfrone G. Work-related noise exposure in a cohort of patients with chronic tinnitus: Analysis of demographic and audiological characteristics. Int J Environ Res Public Health. 2017;14:1035. doi: 10.3390/ijerph14091035. - DOI - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
