

Base editing repairs an SGCA mutation in human primary muscle stem cells
- PMID: 33848270
- PMCID: PMC8262330
- DOI: 10.1172/jci.insight.145994
Base editing repairs an SGCA mutation in human primary muscle stem cells
Abstract
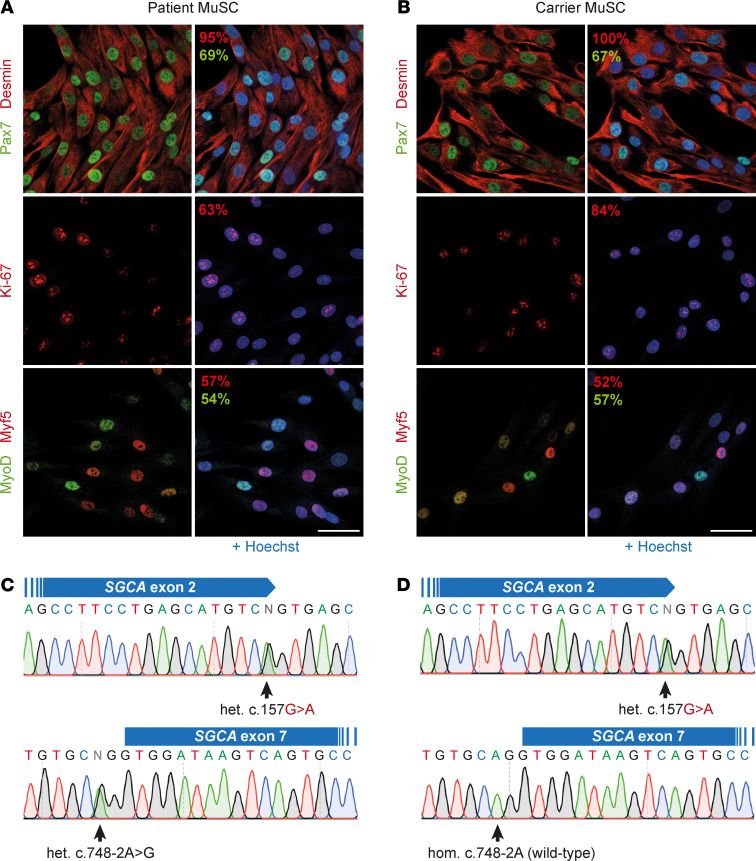
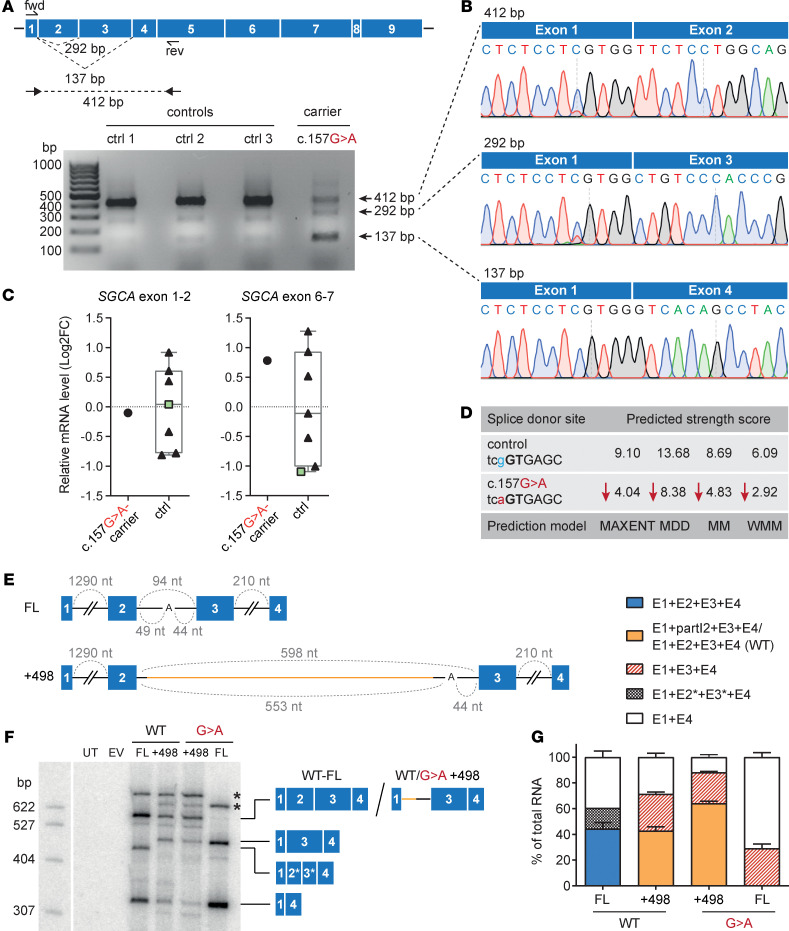
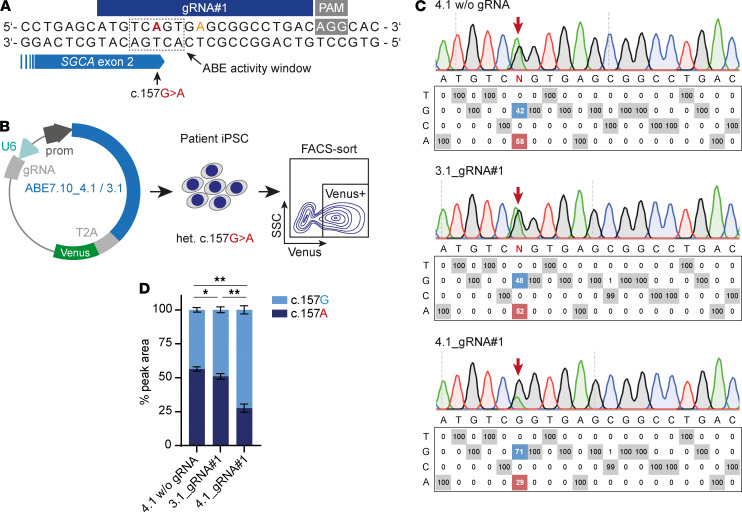
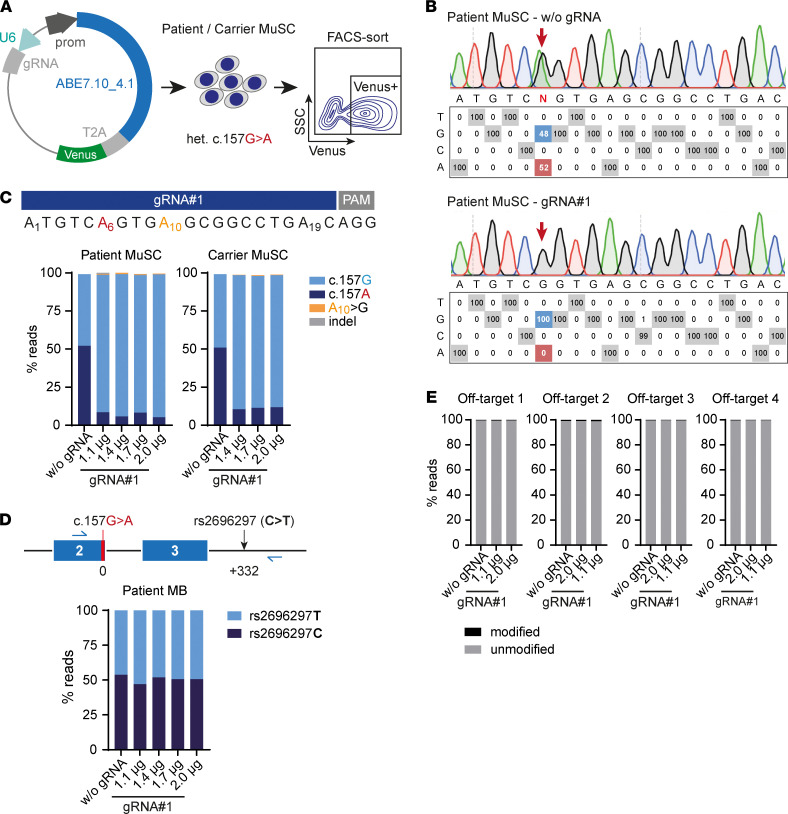
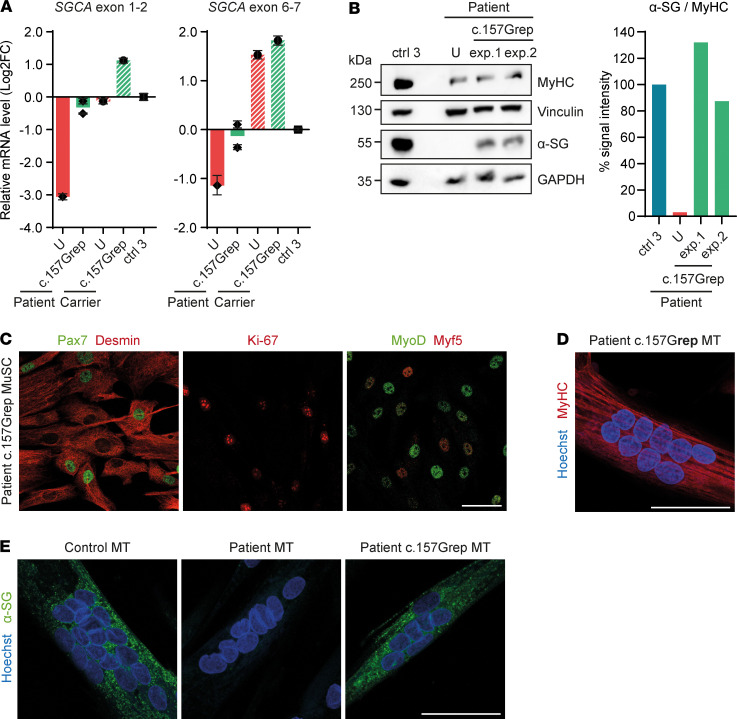
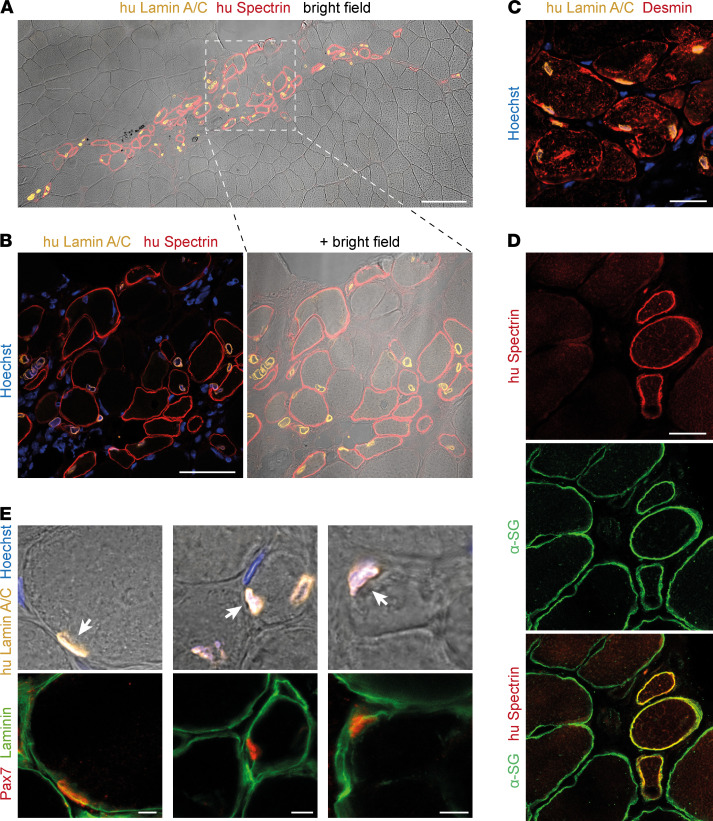
Skeletal muscle can regenerate from muscle stem cells and their myogenic precursor cell progeny, myoblasts. However, precise gene editing in human muscle stem cells for autologous cell replacement therapies of untreatable genetic muscle diseases has not yet been reported. Loss-of-function mutations in SGCA, encoding α-sarcoglycan, cause limb-girdle muscular dystrophy 2D/R3, an early-onset, severe, and rapidly progressive form of muscular dystrophy affecting both male and female patients. Patients suffer from muscle degeneration and atrophy affecting the limbs, respiratory muscles, and heart. We isolated human muscle stem cells from 2 donors, with the common SGCA c.157G>A mutation affecting the last coding nucleotide of exon 2. We found that c.157G>A is an exonic splicing mutation that induces skipping of 2 coregulated exons. Using adenine base editing, we corrected the mutation in the cells from both donors with > 90% efficiency, thereby rescuing the splicing defect and α-sarcoglycan expression. Base-edited patient cells regenerated muscle and contributed to the Pax7+ satellite cell compartment in vivo in mouse xenografts. Here, we provide the first evidence to our knowledge that autologous gene-repaired human muscle stem cells can be harnessed for cell replacement therapies of muscular dystrophies.
Keywords: Human stem cells; Monogenic diseases; Skeletal muscle; Stem cells; Therapeutics.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
